




Shuai.Chen(陈帅)
@schen9.bsky.social
PhD student at UG, researching artificial organic chemistry and inverse molecule design with AI under Robert Pollice.
Reposted by Shuai.Chen(陈帅)

Out now in JACS! We show that under typical Suzuki-Miyaura cross-coupling conditions, bulky ligands can promote palladium-catalyzed protodeboronation.
📜 pubs.acs.org/doi/full/10....
🖥️ github.com/chertianser/...
[1/5]
📜 pubs.acs.org/doi/full/10....
🖥️ github.com/chertianser/...
[1/5]
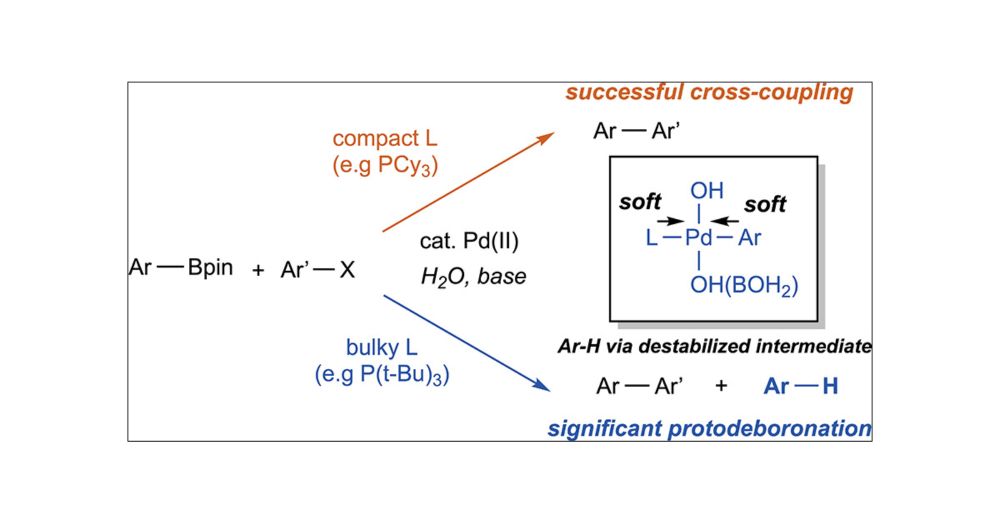
Bulky Phosphine Ligands Promote Palladium-Catalyzed Protodeboronation
The Suzuki–Miyaura cross-coupling reaction is plagued by protodeboronation, an undesirable side reaction with water that consumes the boronic acid derivatives required for the cross-coupling reaction....
pubs.acs.org
November 19, 2025 at 5:48 PM
Out now in JACS! We show that under typical Suzuki-Miyaura cross-coupling conditions, bulky ligands can promote palladium-catalyzed protodeboronation.
📜 pubs.acs.org/doi/full/10....
🖥️ github.com/chertianser/...
[1/5]
📜 pubs.acs.org/doi/full/10....
🖥️ github.com/chertianser/...
[1/5]
Reposted by Shuai.Chen(陈帅)

Rapid generation of transition-state conformer ensembles via constrained distance geometry | ChemRxiv - doi.org/10.26434/che... #compchem

Rapid generation of transition-state conformer ensembles via constrained distance geometry
The consideration of transition state (TS) conformer ensembles is required to accurately model a reaction, and thus plays a key role in computational catalyst design. While CREST and GOAT are establis...
doi.org
November 20, 2025 at 8:45 AM
Rapid generation of transition-state conformer ensembles via constrained distance geometry | ChemRxiv - doi.org/10.26434/che... #compchem
Reposted by Shuai.Chen(陈帅)

#RobSelects preprint 1 of the week #ChemRxiv: Reaching density functional approxmation accuracy at the cost of extended tight-binding quantum chemistry. #compchem https://doi.org/10.26434/chemrxiv-2025-bjxvt

g-xTB: A General-Purpose Extended Tight-Binding Electronic Structure Method For the Elements H to Lr (Z=1–103)
We present g-xTB, a next-generation semi-empirical electronic structure method derived from tight-binding (TB) approximations to Kohn–Sham density functional theory (KS-DFT). Designed to bridge the gap between semi-empirical quantum mechanical (SQM) approaches and DFT in terms of accuracy, robustness, and general applicability, g-xTB targets the performance of the ωB97M-V range-separated hybrid density functional with large basis sets while maintaining TB speed. Key innovations include an atom-in-molecule adaptive atomic orbital basis, a refined Hamiltonian incorporating range-separated approximate Fock exchange, up to fourth-order charge-fluctuation terms with a novel first-order electronic contribution, and atomic correction potentials (ACPs), as well as a charge-dependent semi-classical repulsion function. Parameterized on extended and extremely diverse molecular training sets – including “mindless molecules” – g-xTB achieves excellent accuracy across a broad chemical space, including the actinide elements. Benchmarking against around 32k relative energies across thermochemistry, conformational energetics, non-covalent interactions, and reaction barriers shows that g-xTB consistently outperforms GFN2-xTB, often reducing mean absolute errors by half. Notably, it achieves a WTMAD-2 of 9.3 kcal mol−1 on the full GMTKN55 benchmark, comparable to low-cost DFT methods. It also shows substantial improvements for transition-metal complexes, relative spin state energies, and orbital energy gaps – areas where many SQM and even DFT methods often struggle. In summary, g-xTB offers DFT-like accuracy with minimal computational overhead compared to its predecessor, GFN2-xTB, making it a robust, minimally empirical, transferable, and efficient alternative to machine learning interatomic potentials for a wide range of molecular simulations. It is proposed as a general replacement for the GFNn-xTB family and, in many practical cases, a viable substitute for low- and mid-level DFT methods.
chemrxiv.org
July 1, 2025 at 7:59 PM
#RobSelects preprint 1 of the week #ChemRxiv: Reaching density functional approxmation accuracy at the cost of extended tight-binding quantum chemistry. #compchem https://doi.org/10.26434/chemrxiv-2025-bjxvt
Reposted by Shuai.Chen(陈帅)
#RobSelects preprint of the week #ChemRxiv: Efficient prediction of transition state geometries from molecular strings of starting materials and products via E(3)-equivariant flow-matching. #aichem https://doi.org/10.26434/chemrxiv-2025-bk2rh

GoFlow: Efficient Transition State Geometry Prediction with Flow Matching and E(3)-Equivariant Neural Networks
Transition state (TS) geometries of chemical reactions are key to understanding reaction mechanisms and estimating kinetic properties. Inferring these directly from 2D reaction graphs offers chemists a powerful tool for rapid and accessible reaction analysis. Quantum chemical methods for computing TSs are computationally intensive and often infeasible for larger molecular systems. Recently, deep learning–based diffusion models have shown promise in generating TSs from 2D reaction graphs for single-step reactions. However, framing TS generation as a diffusion process, by design, requires a prohibitively large number of sampling steps during inference. Here we show that modeling TS generation as an optimal transport flow problem, solved via E(3)-equivariant flow matching with geometric tensor networks, achieves over a hundredfold speedup in inference while improving geometric accuracy compared to the state-of-the-art. This breakthrough increase in sampling efficiency and predictive accuracy enables the practical use of deep learning-based TS generators in high-throughput settings for larger and more complex chemical systems. Our method, GoFlow, thus represents a significant methodological advancement in machine learning-based TS generation, bringing it closer to widespread use in computational chemistry workflows.
chemrxiv.org
May 5, 2025 at 11:34 AM
#RobSelects preprint of the week #ChemRxiv: Efficient prediction of transition state geometries from molecular strings of starting materials and products via E(3)-equivariant flow-matching. #aichem https://doi.org/10.26434/chemrxiv-2025-bk2rh
Reposted by Shuai.Chen(陈帅)
#RobSelects preprint of the week #ChemRxiv: Developing an enantioselectivity prediction workflow for asymmetric Sharpless dihydroxylation. #catalysis https://doi.org/10.26434/chemrxiv-2025-zp7rn

Data-Driven Prediction of Enantioselectivity for the Sharpless Asymmetric Dihydroxylation: Model Development and Experimental Validation
The Sharpless asymmetric dihydroxylation remains a key transformation in chemical synthesis, yet its success hides unexpected cases of lower selectivity. A chemoinformatic workflow was developed to allow data-driven analysis of the reaction. A database of 1007 reactions employing AD-mix α and β was curated from the literature, and an alignment-dependent, fragment-based featurization of alkenes was implemented for modeling. This platform converged on machine learning models capable of predicting the magnitude of enantioselectivity for multiple alkene classes, achieving Q2F3 values ≥ 0.8, test r2 values ≥ 0.7 and mean absolute errors (MAE) ≤ 0.3 kcal/mol. The features of alkenes contributing to model performance were assessed with SHapley Additive exPlanations (SHAP) analysis to gather insight into factors underlying predictions. Experimental validation demonstrated that the models could achieve meaningful predictions on numerous out-of-sample alkenes.
chemrxiv.org
April 28, 2025 at 8:18 AM
#RobSelects preprint of the week #ChemRxiv: Developing an enantioselectivity prediction workflow for asymmetric Sharpless dihydroxylation. #catalysis https://doi.org/10.26434/chemrxiv-2025-zp7rn
Reposted by Shuai.Chen(陈帅)

Our latest ChemRxiv preprint, "Transferable #MachineLearning Interatomic Potential for Pd-Catalyzed Cross-Coupling Reactions" Collaboration with @nsf-ccas.bsky.social @gabegomes.bsky.social @bobbypaton.bsky.social chemrxiv.org/engage/chemr... #compchem #chemsky
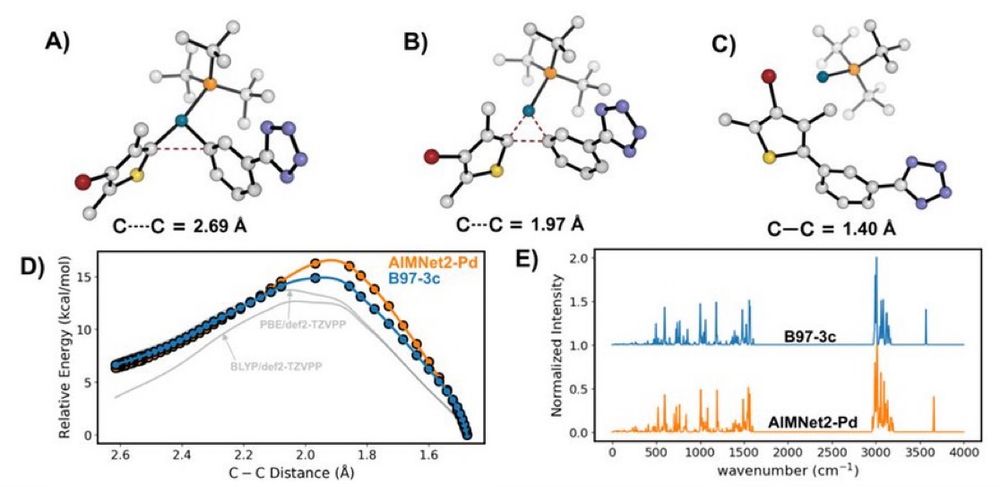
March 18, 2025 at 2:12 PM
Our latest ChemRxiv preprint, "Transferable #MachineLearning Interatomic Potential for Pd-Catalyzed Cross-Coupling Reactions" Collaboration with @nsf-ccas.bsky.social @gabegomes.bsky.social @bobbypaton.bsky.social chemrxiv.org/engage/chemr... #compchem #chemsky
Reposted by Shuai.Chen(陈帅)
#RobSelects paper of the week #j_a_c_s: Photochemical manganese- or rhenium-catalyzed carbon-hydrogen activation of methane and benzene to form carbon-beryllium bonds. #catalysis https://doi.org/10.1021/jacs.5c02179
March 17, 2025 at 8:46 PM
#RobSelects paper of the week #j_a_c_s: Photochemical manganese- or rhenium-catalyzed carbon-hydrogen activation of methane and benzene to form carbon-beryllium bonds. #catalysis https://doi.org/10.1021/jacs.5c02179
Reposted by Shuai.Chen(陈帅)

More mobile lab robots from the team at USTC, China, this time integrated using LLMs, just out in @jamchemsoc.bsky.social: pubs.acs.org/doi/epdf/10....

A Multiagent-Driven Robotic AI Chemist Enabling Autonomous Chemical Research On Demand
The successful integration of large language models (LLMs) into laboratory workflows has demonstrated robust capabilities in natural language processing, autonomous task execution, and collaborative p...
pubs.acs.org
March 10, 2025 at 12:49 PM
More mobile lab robots from the team at USTC, China, this time integrated using LLMs, just out in @jamchemsoc.bsky.social: pubs.acs.org/doi/epdf/10....
Reposted by Shuai.Chen(陈帅)

Really grateful to be part of this review on predictions of selectivity, driven by @lukasmsigmund.bsky.social ! doi.org/10.1039/D5SC...
Computational tools for the prediction of site- and regioselectivity of organic reactions
The regio- and site-selectivity of organic reactions is one of the most important aspects when it comes to synthesis planning. Due to that, massive research efforts were invested into computational mo...
doi.org
March 10, 2025 at 4:34 PM
Really grateful to be part of this review on predictions of selectivity, driven by @lukasmsigmund.bsky.social ! doi.org/10.1039/D5SC...
Reposted by Shuai.Chen(陈帅)

Now online & open access:
Article by R Güdük, N Kehl, C Stavagna, MJ. Tilby, O Turner, A Ruffoni, HP. Caldora & D Leonori
A three-step strategy for the conversion of pyridines into benzonitriles
www.nature.com/articles/s44...
#ChemSky
Article by R Güdük, N Kehl, C Stavagna, MJ. Tilby, O Turner, A Ruffoni, HP. Caldora & D Leonori
A three-step strategy for the conversion of pyridines into benzonitriles
www.nature.com/articles/s44...
#ChemSky

A three-step strategy for the conversion of pyridines into benzonitriles - Nature Synthesis
Bioisosteric replacement is vital in drug discovery; however, substituting core ring structures is challenging. Now, a strategy that converts pyridines into benzonitriles, via N-oxidation, photochemic...
www.nature.com
March 7, 2025 at 10:21 AM
Now online & open access:
Article by R Güdük, N Kehl, C Stavagna, MJ. Tilby, O Turner, A Ruffoni, HP. Caldora & D Leonori
A three-step strategy for the conversion of pyridines into benzonitriles
www.nature.com/articles/s44...
#ChemSky
Article by R Güdük, N Kehl, C Stavagna, MJ. Tilby, O Turner, A Ruffoni, HP. Caldora & D Leonori
A three-step strategy for the conversion of pyridines into benzonitriles
www.nature.com/articles/s44...
#ChemSky
Reposted by Shuai.Chen(陈帅)

New online! Enantioselective C–H annulations enabled by either nickel- or cobalt-electrocatalysed C–H activation for catalyst-controlled chemodivergence

Enantioselective C–H annulations enabled by either nickel- or cobalt-electrocatalysed C–H activation for catalyst-controlled chemodivergence
Nature Catalysis, Published online: 07 March 2025; doi:10.1038/s41929-025-01306-9Controlling the selectivity of electrocatalysed C–H activation with earth-abundant base metals can increase its synthetic impact. Now chemodivergence of an electrocatalysed…
bit.ly
March 7, 2025 at 8:29 PM
New online! Enantioselective C–H annulations enabled by either nickel- or cobalt-electrocatalysed C–H activation for catalyst-controlled chemodivergence
Reposted by Shuai.Chen(陈帅)

🚀 Looking for reaction conditions that work well for multiple substrates? CurryBO can help🍛
Now out on arXiv: arxiv.org/abs/2502.18966
A short explanation thread 👇
Now out on arXiv: arxiv.org/abs/2502.18966
A short explanation thread 👇

One Set to Rule Them All: How to Obtain General Chemical Conditions via Bayesian Optimization over Curried Functions
General parameters are highly desirable in the natural sciences - e.g., chemical reaction conditions that enable high yields across a range of related transformations. This has a significant practical...
arxiv.org
March 3, 2025 at 9:06 AM
🚀 Looking for reaction conditions that work well for multiple substrates? CurryBO can help🍛
Now out on arXiv: arxiv.org/abs/2502.18966
A short explanation thread 👇
Now out on arXiv: arxiv.org/abs/2502.18966
A short explanation thread 👇
Reposted by Shuai.Chen(陈帅)

This is old article but worth to read.
pubmed.ncbi.nlm.nih.gov/25820774/
pubmed.ncbi.nlm.nih.gov/25820774/

How many molecules does it take to tell a story? Case studies, language, and an epistemic view of medicinal chemistry - PubMed
Medicinal chemistry has always been closer to the arts than other disciplines in the natural sciences. Instead of searching for natural laws, medicinal chemistry creates new molecular entities entaili...
pubmed.ncbi.nlm.nih.gov
February 23, 2025 at 11:29 AM
This is old article but worth to read.
pubmed.ncbi.nlm.nih.gov/25820774/
pubmed.ncbi.nlm.nih.gov/25820774/
Reposted by Shuai.Chen(陈帅)

We start our first post on Bluesky with a firework! Very proud of a brilliant team to publish our work on C(sp3)-atom transfer @science.org. www.science.org/doi/10.1126/... It has been a very exciting journey. Thanks @erc.europa.eu
February 20, 2025 at 7:42 PM
We start our first post on Bluesky with a firework! Very proud of a brilliant team to publish our work on C(sp3)-atom transfer @science.org. www.science.org/doi/10.1126/... It has been a very exciting journey. Thanks @erc.europa.eu