




Martin Steinegger 🇺🇦
@martinsteinegger.bsky.social
Developing data intensive computational methods • PI @ Seoul National University 🇰🇷 • #FirstGen • he/him • Hauptschüler
End-to-end protein design in the browser through evedesign. Generate and interactively explore designs in 2D/3D and export them as codon-optimized DNA. The underlying open source framework (released soon) is build to easily add new methods, more on that soon.
🌐 evedesign.bio
🌐 evedesign.bio
October 22, 2025 at 2:30 PM
End-to-end protein design in the browser through evedesign. Generate and interactively explore designs in 2D/3D and export them as codon-optimized DNA. The underlying open source framework (released soon) is build to easily add new methods, more on that soon.
🌐 evedesign.bio
🌐 evedesign.bio
Below we show GPU-accelerated Foldseek, searching 128 structures against AFDB50 (54 million structures). On 128 CPU cores this takes ~120 seconds, whereas a single GPU completes it in ~25 seconds. 2/n
September 21, 2025 at 8:06 AM
Below we show GPU-accelerated Foldseek, searching 128 structures against AFDB50 (54 million structures). On 128 CPU cores this takes ~120 seconds, whereas a single GPU completes it in ~25 seconds. 2/n
@pedrobeltrao.bsky.social here you can see the clustering at 90% identity.

September 3, 2025 at 9:53 AM
@pedrobeltrao.bsky.social here you can see the clustering at 90% identity.
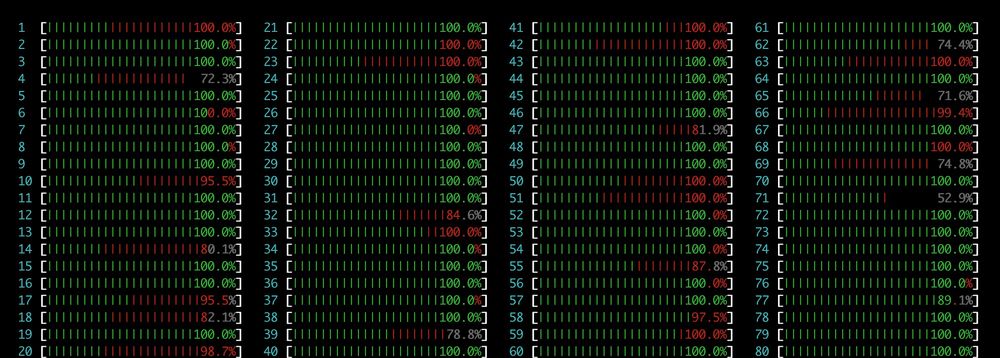
If you use Boltz1/2, BioEmu, Chai1, or other MSA-dependent models, you’re likely using our ColabFold server. Please be considerate! Avoid large submissions across many IPs instead generate the MSA locally. Our server is an old-timer from 2014 and can’t handle that load.

August 15, 2025 at 5:48 PM
If you use Boltz1/2, BioEmu, Chai1, or other MSA-dependent models, you’re likely using our ColabFold server. Please be considerate! Avoid large submissions across many IPs instead generate the MSA locally. Our server is an old-timer from 2014 and can’t handle that load.
MMseqs2 v18 is out
- SIMD FW/BW alignment (preprint soon!)
- Sub. Mat. λ calculator by Eric Dawson
- Faster ARM SW by Alexander Nesterovskiy
- MSA-Pairformer’s proximity-based pairing for multimer prediction (www.biorxiv.org/content/10.1...; avail. in ColabFold API)
💾 github.com/soedinglab/M... & 🐍
- SIMD FW/BW alignment (preprint soon!)
- Sub. Mat. λ calculator by Eric Dawson
- Faster ARM SW by Alexander Nesterovskiy
- MSA-Pairformer’s proximity-based pairing for multimer prediction (www.biorxiv.org/content/10.1...; avail. in ColabFold API)
💾 github.com/soedinglab/M... & 🐍

August 5, 2025 at 8:25 AM
MMseqs2 v18 is out
- SIMD FW/BW alignment (preprint soon!)
- Sub. Mat. λ calculator by Eric Dawson
- Faster ARM SW by Alexander Nesterovskiy
- MSA-Pairformer’s proximity-based pairing for multimer prediction (www.biorxiv.org/content/10.1...; avail. in ColabFold API)
💾 github.com/soedinglab/M... & 🐍
- SIMD FW/BW alignment (preprint soon!)
- Sub. Mat. λ calculator by Eric Dawson
- Faster ARM SW by Alexander Nesterovskiy
- MSA-Pairformer’s proximity-based pairing for multimer prediction (www.biorxiv.org/content/10.1...; avail. in ColabFold API)
💾 github.com/soedinglab/M... & 🐍
Folddisco webserver result view update:
- Added description texts for AFDB
- Integrated TaxoView taxonomy visualization & filter by @sunjaelee.bsky.social
- Inter-residue distance clustering by DBSCAN to explore motif diversity.
🌐 search.foldseek.com/folddisco
📄 www.biorxiv.org/content/10.1...
- Added description texts for AFDB
- Integrated TaxoView taxonomy visualization & filter by @sunjaelee.bsky.social
- Inter-residue distance clustering by DBSCAN to explore motif diversity.
🌐 search.foldseek.com/folddisco
📄 www.biorxiv.org/content/10.1...
July 22, 2025 at 6:11 PM
Folddisco webserver result view update:
- Added description texts for AFDB
- Integrated TaxoView taxonomy visualization & filter by @sunjaelee.bsky.social
- Inter-residue distance clustering by DBSCAN to explore motif diversity.
🌐 search.foldseek.com/folddisco
📄 www.biorxiv.org/content/10.1...
- Added description texts for AFDB
- Integrated TaxoView taxonomy visualization & filter by @sunjaelee.bsky.social
- Inter-residue distance clustering by DBSCAN to explore motif diversity.
🌐 search.foldseek.com/folddisco
📄 www.biorxiv.org/content/10.1...
Today at 5pm, @eunbelivable.bsky.social will present her work on the Big Fantastic Viral Database (BFVD) at #ISMB2025 in BOSC. She also has a poster B-123 (tomorrow, 22nd), so please drop by to have ta chat and grab some stickers!
📄 academic.oup.com/nar/article/...
📄 academic.oup.com/nar/article/...


July 21, 2025 at 9:29 AM
Today at 5pm, @eunbelivable.bsky.social will present her work on the Big Fantastic Viral Database (BFVD) at #ISMB2025 in BOSC. She also has a poster B-123 (tomorrow, 22nd), so please drop by to have ta chat and grab some stickers!
📄 academic.oup.com/nar/article/...
📄 academic.oup.com/nar/article/...
We provide a user-friendly Folddisco webserver, enabling instant structural motif searches in PDB, AFDB-Proteomes, AFDB50 (available later today), and ESMatlas (ESM30). Explore it here: search.foldseek.com/folddisco 8/9
July 7, 2025 at 8:21 AM
We provide a user-friendly Folddisco webserver, enabling instant structural motif searches in PDB, AFDB-Proteomes, AFDB50 (available later today), and ESMatlas (ESM30). Explore it here: search.foldseek.com/folddisco 8/9
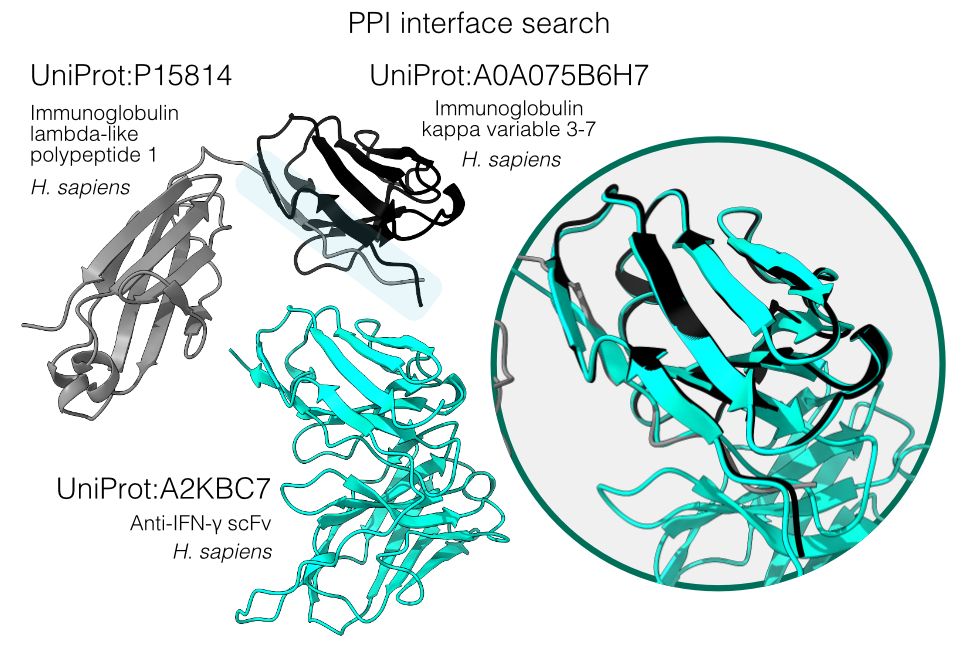
Folddisco can be applied for PPI interface searches. When querying an interface between antibody chains (gray/black), it successfully identifies matching interfaces within monomeric antibody fragments (cyan), showcasing its potential to detect novel interaction partners and interfaces. 7/9
July 7, 2025 at 8:21 AM
Folddisco can be applied for PPI interface searches. When querying an interface between antibody chains (gray/black), it successfully identifies matching interfaces within monomeric antibody fragments (cyan), showcasing its potential to detect novel interaction partners and interfaces. 7/9
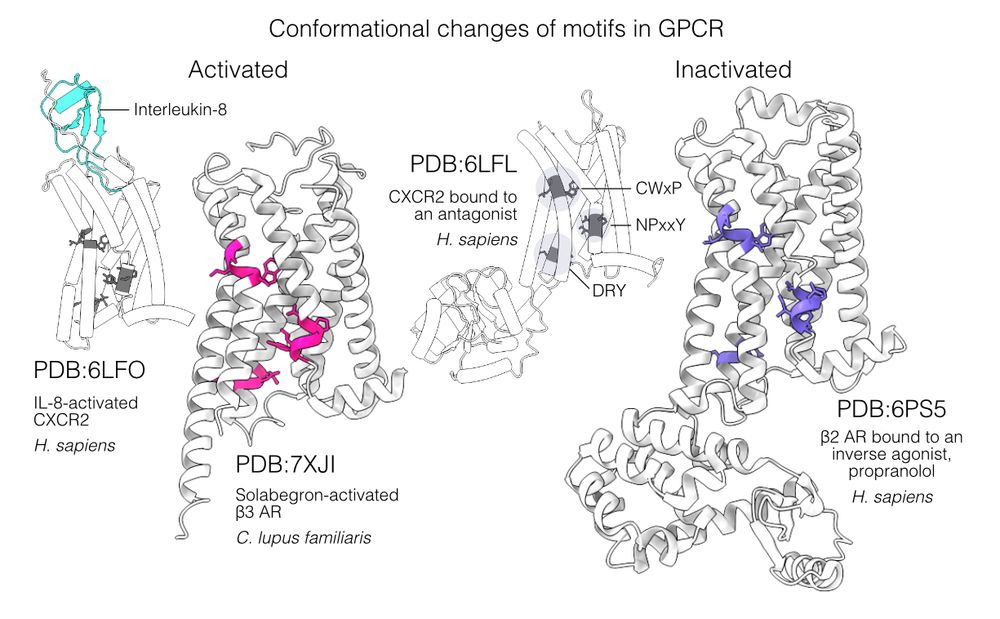
Folddisco can distinguish functional states. We searched GPCR activation motifs (CWxP, NPxxY, DRY), clearly separating active/inactive states. A search in the AFDB shows ~53% active, closely mirroring experimental PDB 54%, suggesting AlphaFold might follow its training conformation distribution. 6/9
July 7, 2025 at 8:21 AM
Folddisco can distinguish functional states. We searched GPCR activation motifs (CWxP, NPxxY, DRY), clearly separating active/inactive states. A search in the AFDB shows ~53% active, closely mirroring experimental PDB 54%, suggesting AlphaFold might follow its training conformation distribution. 6/9
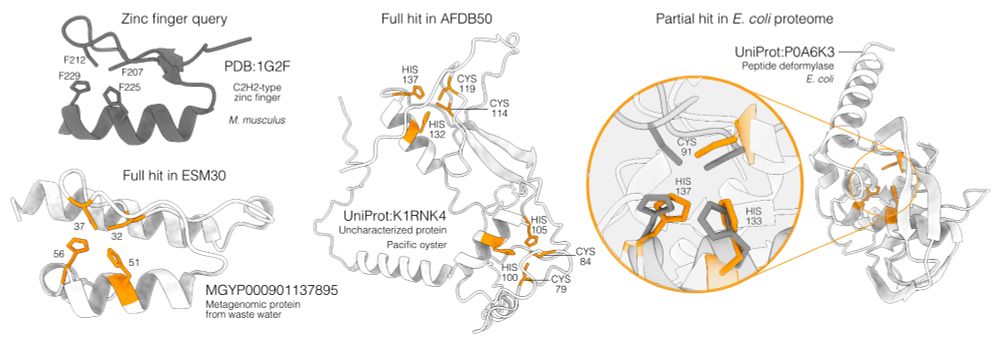
Folddisco can annotate proteins: querying a canonical zinc-finger uncovers an uncharacterized oyster protein and metagenomic proteins. It also detects partial catalytic metal sites in E. coli peptide deformylase. All of these hits would be missed by Foldseek or sequence aligners. 5/9
July 7, 2025 at 8:21 AM
Folddisco can annotate proteins: querying a canonical zinc-finger uncovers an uncharacterized oyster protein and metagenomic proteins. It also detects partial catalytic metal sites in E. coli peptide deformylase. All of these hits would be missed by Foldseek or sequence aligners. 5/9
Folddisco builds indexes faster and smaller than previous tools: indexing AFDB50 (53M structures) takes only ~24h vs. ~20 days (extrapolated) for pyScoMotif. Querying a zinc-finger motif across AFDB50 takes just ~13s, up to 48x faster than pyScoMotif. 4/9
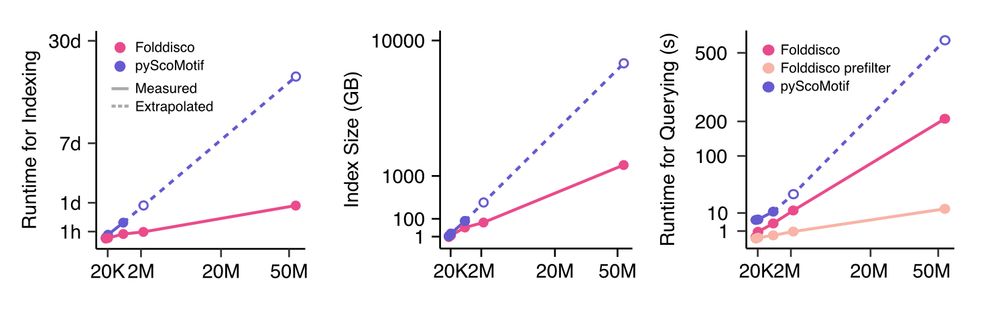
July 7, 2025 at 8:21 AM
Folddisco builds indexes faster and smaller than previous tools: indexing AFDB50 (53M structures) takes only ~24h vs. ~20 days (extrapolated) for pyScoMotif. Querying a zinc-finger motif across AFDB50 takes just ~13s, up to 48x faster than pyScoMotif. 4/9
Folddisco accurately detects discontinuous motifs like zinc fingers and segment-based motifs, previously requiring separate tools. Additionally, we built a SCOPe benchmark by sampling conserved residues from families and measuring the recall up to the first false positive. 3/9
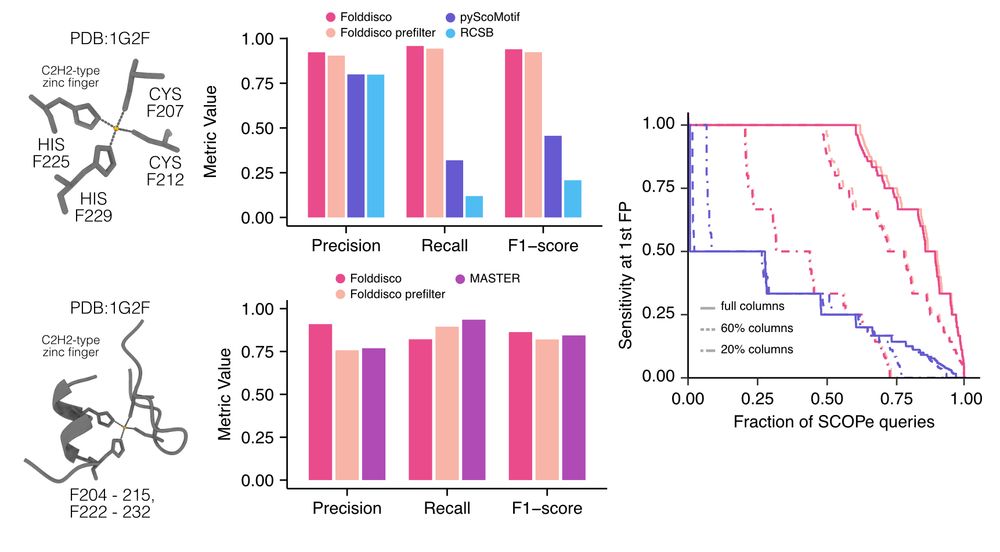
July 7, 2025 at 8:21 AM
Folddisco accurately detects discontinuous motifs like zinc fingers and segment-based motifs, previously requiring separate tools. Additionally, we built a SCOPe benchmark by sampling conserved residues from families and measuring the recall up to the first false positive. 3/9
Folddisco detects (partial) motifs, allowing for substitutions and angle-length variations, by utilizing an index storing all residue pairs within 20Å encoded as geometric features. For space efficiency, it omits positions and compresses ids through run-length encoding (1.4TB for 53M structures) 2/9
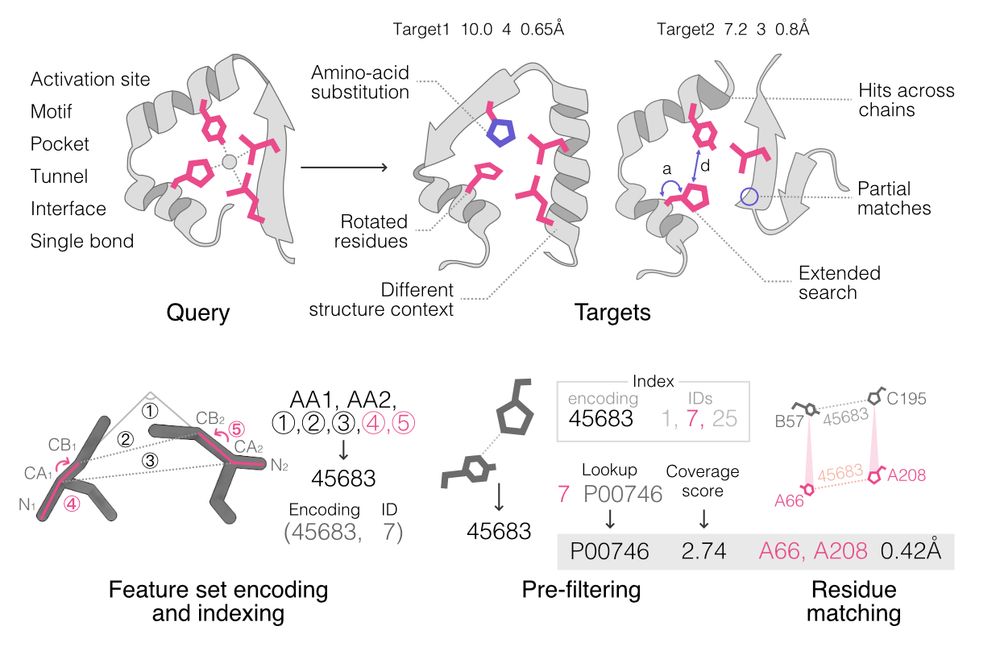
July 7, 2025 at 8:21 AM
Folddisco detects (partial) motifs, allowing for substitutions and angle-length variations, by utilizing an index storing all residue pairs within 20Å encoded as geometric features. For space efficiency, it omits positions and compresses ids through run-length encoding (1.4TB for 53M structures) 2/9
Folddisco finds similar (dis)continuous 3D motifs in large protein structure databases. Its efficient index enables fast uncharacterized active site annotation, protein conformational state analysis and PPI interface comparison. 1/9🧶🧬
📄 www.biorxiv.org/content/10.1...
🌐 search.foldseek.com/folddisco
📄 www.biorxiv.org/content/10.1...
🌐 search.foldseek.com/folddisco
July 7, 2025 at 8:21 AM
Folddisco finds similar (dis)continuous 3D motifs in large protein structure databases. Its efficient index enables fast uncharacterized active site annotation, protein conformational state analysis and PPI interface comparison. 1/9🧶🧬
📄 www.biorxiv.org/content/10.1...
🌐 search.foldseek.com/folddisco
📄 www.biorxiv.org/content/10.1...
🌐 search.foldseek.com/folddisco
We've updated our AFESM website to now include biome filtering, allowing exploration of protein structures adapted to specific environments.
🌐 afesm.foldseek.com
Read more about the work in the skeetorial
🦋 bsky.app/profile/mart...
or our preprint
📄 www.biorxiv.org/content/10.1...
🌐 afesm.foldseek.com
Read more about the work in the skeetorial
🦋 bsky.app/profile/mart...
or our preprint
📄 www.biorxiv.org/content/10.1...
May 15, 2025 at 2:03 PM
We've updated our AFESM website to now include biome filtering, allowing exploration of protein structures adapted to specific environments.
🌐 afesm.foldseek.com
Read more about the work in the skeetorial
🦋 bsky.app/profile/mart...
or our preprint
📄 www.biorxiv.org/content/10.1...
🌐 afesm.foldseek.com
Read more about the work in the skeetorial
🦋 bsky.app/profile/mart...
or our preprint
📄 www.biorxiv.org/content/10.1...
My student build a huge version of it! :D

May 15, 2025 at 8:43 AM
My student build a huge version of it! :D
First assembled version

May 12, 2025 at 7:43 AM
First assembled version
ESMfold struggles to predict ~7k novel TED-detected AFDB domains, showing consistently lower pLDDT (Suppl. Fig. 11). For the ESMatlas, we did re-predict ~25% of ESM-only clusters with AF2 (ColabFold) and detected only one novel fold.

April 30, 2025 at 9:39 AM
ESMfold struggles to predict ~7k novel TED-detected AFDB domains, showing consistently lower pLDDT (Suppl. Fig. 11). For the ESMatlas, we did re-predict ~25% of ESM-only clusters with AF2 (ColabFold) and detected only one novel fold.
Explore AFESM with our website! You can search your favorite proteins from ESMatlas or AFDB using their identifiers. It's still a work in progress, with many exciting features on the way! 7/n
🌐 afesm.foldseek.com
🌐 afesm.foldseek.com
April 27, 2025 at 12:13 AM
Explore AFESM with our website! You can search your favorite proteins from ESMatlas or AFDB using their identifiers. It's still a work in progress, with many exciting features on the way! 7/n
🌐 afesm.foldseek.com
🌐 afesm.foldseek.com
However, these novel domain combinations explain only a small fraction (0.3%) of the ESM-only clusters. The remainder are mostly low-quality predictions (53%), fragments (16%), known domains with potential unknown extensions (19%), or without identifiable domains (9.3%). 6/n
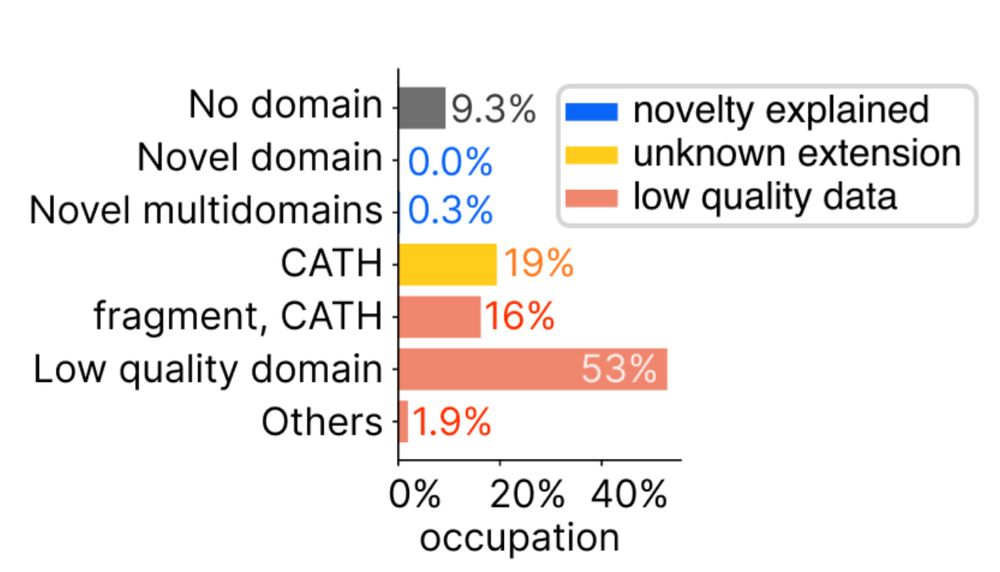
April 27, 2025 at 12:13 AM
However, these novel domain combinations explain only a small fraction (0.3%) of the ESM-only clusters. The remainder are mostly low-quality predictions (53%), fragments (16%), known domains with potential unknown extensions (19%), or without identifiable domains (9.3%). 6/n
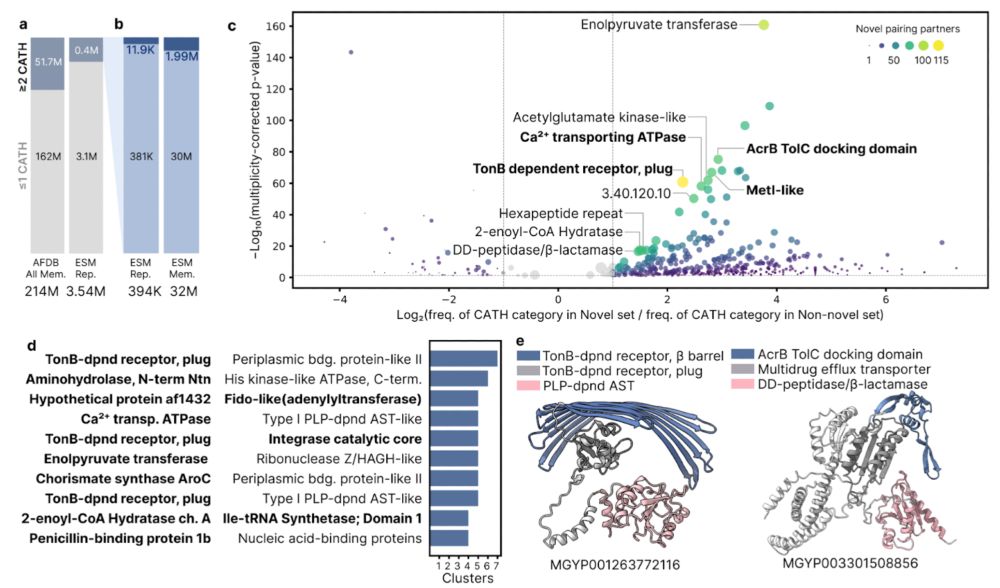
We identified 11,941 novel multi-domain combinations. We found membrane-associated domains (e.g., TonB dependent receptor, highlighting domain recombination rather than new folds as a driver of structural innovation. 5/n
April 27, 2025 at 12:13 AM
We identified 11,941 novel multi-domain combinations. We found membrane-associated domains (e.g., TonB dependent receptor, highlighting domain recombination rather than new folds as a driver of structural innovation. 5/n
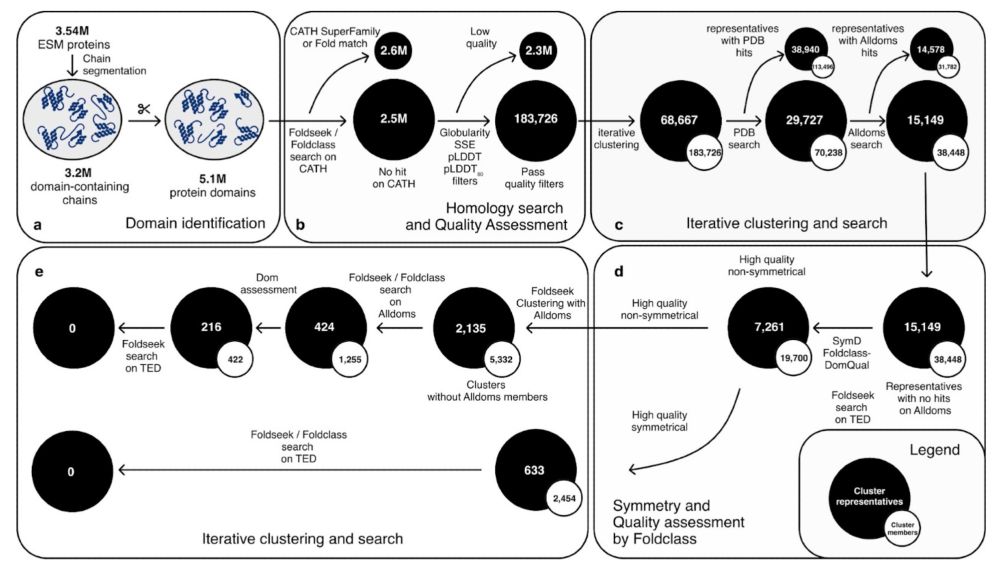
We annotated domains in ESM-only clusters using the TED workflow and found 0 new folds. Re-modelling ~25% of proteins with AlphaFold2 revealed 1 novel fold; unlike AFDB’s >7k novel folds surprisingly, hinting at a saturating fold space or predictor limits.
📄 doi.org/10.1126/scie... 4/n
📄 doi.org/10.1126/scie... 4/n
April 27, 2025 at 12:13 AM
We annotated domains in ESM-only clusters using the TED workflow and found 0 new folds. Re-modelling ~25% of proteins with AlphaFold2 revealed 1 novel fold; unlike AFDB’s >7k novel folds surprisingly, hinting at a saturating fold space or predictor limits.
📄 doi.org/10.1126/scie... 4/n
📄 doi.org/10.1126/scie... 4/n
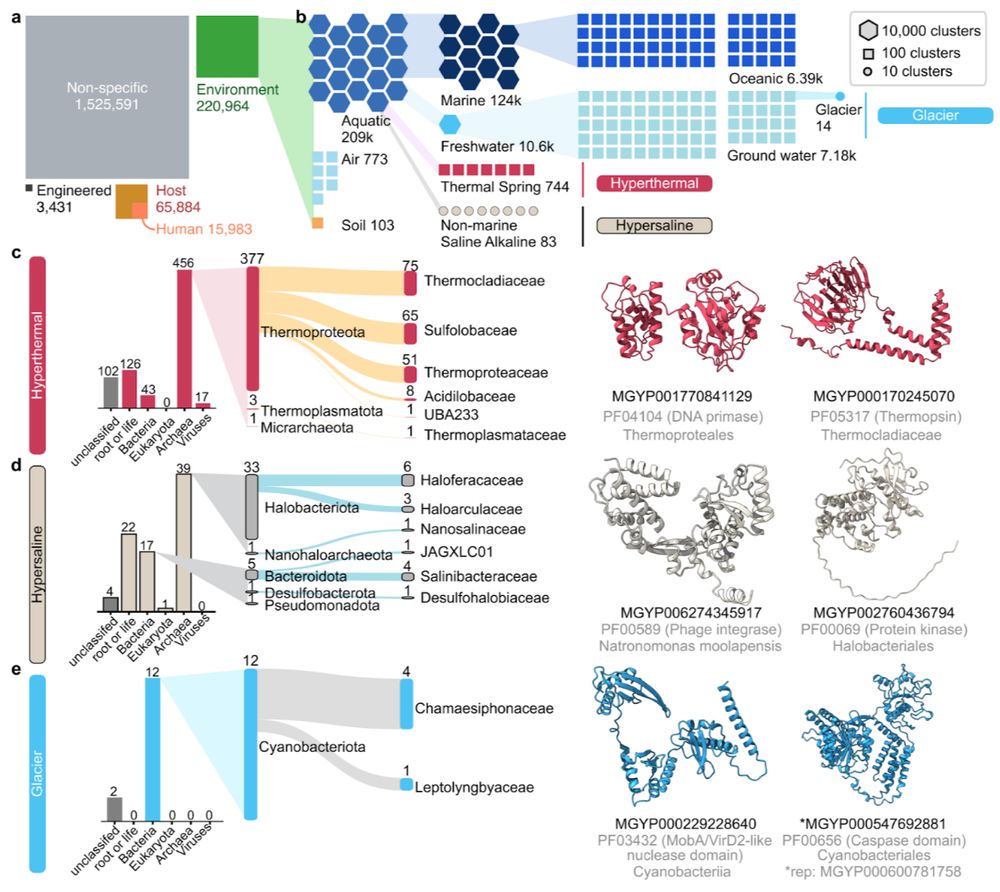
ESMatlas uses MGnify environmental labels. Leveraging this, we computed the lowest common biomes per structural cluster, revealing protein adaptations unique to specific environments, especially extreme ones like hyperthermal, hypersaline, and glaciers. 3/n
April 27, 2025 at 12:13 AM
ESMatlas uses MGnify environmental labels. Leveraging this, we computed the lowest common biomes per structural cluster, revealing protein adaptations unique to specific environments, especially extreme ones like hyperthermal, hypersaline, and glaciers. 3/n
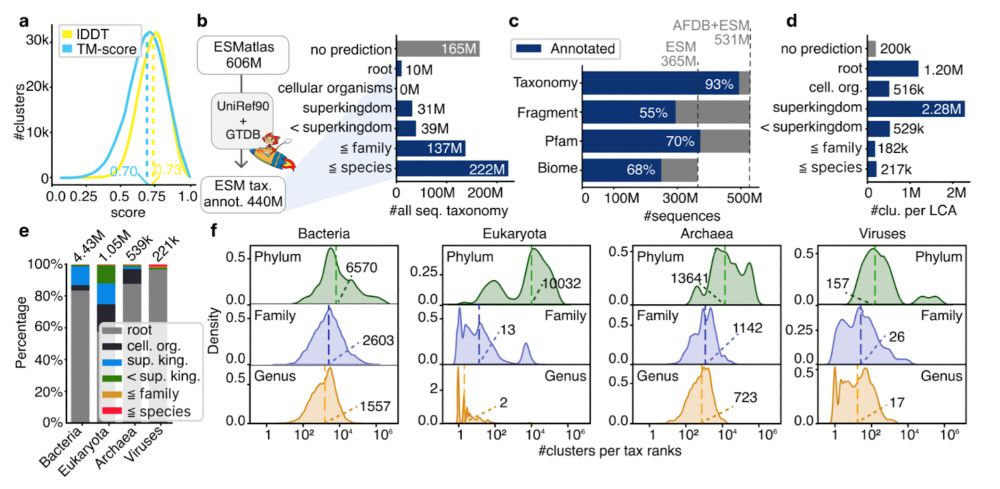
We annotated all ESMatlas entries with MMseqs2 taxonomy (93% coverage) and computed lowest common ancestors (LCA). Most LCAs at superkingdom level, indicating structures shared across domains. Avg. clusters/genus: Bacteria 1,557; Archaea 723; Viruses 17; Eukaryotes 2 (sampling bias). 2/n
April 27, 2025 at 12:13 AM
We annotated all ESMatlas entries with MMseqs2 taxonomy (93% coverage) and computed lowest common ancestors (LCA). Most LCAs at superkingdom level, indicating structures shared across domains. Avg. clusters/genus: Bacteria 1,557; Archaea 723; Viruses 17; Eukaryotes 2 (sampling bias). 2/n