



Jim Shaw
@jimshaw.bsky.social
Postdoc at Dana-Farber and Harvard Med with Heng Li (@lh3lh3.bsky.social). Prev: UBC / UofT.
I like thinking about computational biological sequence analysis and its applications to metagenomics.
https://jim-shaw-bluenote.github.io
I like thinking about computational biological sequence analysis and its applications to metagenomics.
https://jim-shaw-bluenote.github.io
On a public oral ONT metagenome (from @ykiguchi.bsky.social), we assembled a lot more complete, similar (within species-level) genomes than previous methods.
So much to explore... for example, we compared 6 circular TM7 bacteria of > 93% ANI assembled from a single oral metagenome.
10 / N
So much to explore... for example, we compared 6 circular TM7 bacteria of > 93% ANI assembled from a single oral metagenome.
10 / N

September 7, 2025 at 11:35 PM
On a public oral ONT metagenome (from @ykiguchi.bsky.social), we assembled a lot more complete, similar (within species-level) genomes than previous methods.
So much to explore... for example, we compared 6 circular TM7 bacteria of > 93% ANI assembled from a single oral metagenome.
10 / N
So much to explore... for example, we compared 6 circular TM7 bacteria of > 93% ANI assembled from a single oral metagenome.
10 / N
For this gut sample, @mgmarin.bsky.social found two distinct ermF (erythromycin resistance) genes, with 98% similarity, spreading within Bacteroidota.
1. The distinct ermFs are spreading on two distinct MGEs.
2. There is even strain specificity, only 1/6 P. copri had it!
9 / N
1. The distinct ermFs are spreading on two distinct MGEs.
2. There is even strain specificity, only 1/6 P. copri had it!
9 / N

September 7, 2025 at 11:35 PM
For this gut sample, @mgmarin.bsky.social found two distinct ermF (erythromycin resistance) genes, with 98% similarity, spreading within Bacteroidota.
1. The distinct ermFs are spreading on two distinct MGEs.
2. There is even strain specificity, only 1/6 P. copri had it!
9 / N
1. The distinct ermFs are spreading on two distinct MGEs.
2. There is even strain specificity, only 1/6 P. copri had it!
9 / N
With circular contigs, we can confidently analyze presence / absence of "stuff" within contigs _without worrying about binning issues_ (as much).
For example, mobile genetic elements, AMR genes that are hard to bin and assemble with short reads...?
8/N
For example, mobile genetic elements, AMR genes that are hard to bin and assemble with short reads...?
8/N
September 7, 2025 at 11:35 PM
With circular contigs, we can confidently analyze presence / absence of "stuff" within contigs _without worrying about binning issues_ (as much).
For example, mobile genetic elements, AMR genes that are hard to bin and assemble with short reads...?
8/N
For example, mobile genetic elements, AMR genes that are hard to bin and assemble with short reads...?
8/N
Does myloasm offer better insights, not just good benchmarks?
It turns out myloasm can recover more near-complete contigs than other ONT methods.
For a gut sample, it could assemble _6 different Prevotella copri genomes_ into single contigs, whereas other methods struggled.
7 / N
It turns out myloasm can recover more near-complete contigs than other ONT methods.
For a gut sample, it could assemble _6 different Prevotella copri genomes_ into single contigs, whereas other methods struggled.
7 / N

September 7, 2025 at 11:35 PM
Does myloasm offer better insights, not just good benchmarks?
It turns out myloasm can recover more near-complete contigs than other ONT methods.
For a gut sample, it could assemble _6 different Prevotella copri genomes_ into single contigs, whereas other methods struggled.
7 / N
It turns out myloasm can recover more near-complete contigs than other ONT methods.
For a gut sample, it could assemble _6 different Prevotella copri genomes_ into single contigs, whereas other methods struggled.
7 / N
My favourite result:
For jointly-sequenced gut samples (thanks to public data from @jjminich.bsky.social), ONT can assemble _more_ circular contigs than HiFi.
This is thanks to ~3-5x increases in circular contigs relative to previous methods.
5 / N
For jointly-sequenced gut samples (thanks to public data from @jjminich.bsky.social), ONT can assemble _more_ circular contigs than HiFi.
This is thanks to ~3-5x increases in circular contigs relative to previous methods.
5 / N

September 7, 2025 at 11:35 PM
My favourite result:
For jointly-sequenced gut samples (thanks to public data from @jjminich.bsky.social), ONT can assemble _more_ circular contigs than HiFi.
This is thanks to ~3-5x increases in circular contigs relative to previous methods.
5 / N
For jointly-sequenced gut samples (thanks to public data from @jjminich.bsky.social), ONT can assemble _more_ circular contigs than HiFi.
This is thanks to ~3-5x increases in circular contigs relative to previous methods.
5 / N
Myloasm seems to have nice results for metagenome-assembled genomes (MAGs).
For ONT R10.4 data, myloasm stands out, especially for retrieving circular, complete contigs.
For HiFi, metaMDBG is very competitive. hifiasm-meta is still strong for gut samples. 4 / N
For ONT R10.4 data, myloasm stands out, especially for retrieving circular, complete contigs.
For HiFi, metaMDBG is very competitive. hifiasm-meta is still strong for gut samples. 4 / N

September 7, 2025 at 11:35 PM
Myloasm seems to have nice results for metagenome-assembled genomes (MAGs).
For ONT R10.4 data, myloasm stands out, especially for retrieving circular, complete contigs.
For HiFi, metaMDBG is very competitive. hifiasm-meta is still strong for gut samples. 4 / N
For ONT R10.4 data, myloasm stands out, especially for retrieving circular, complete contigs.
For HiFi, metaMDBG is very competitive. hifiasm-meta is still strong for gut samples. 4 / N
We do a bunch more assembly graph cleaning using coverages (and even some _theory_!). See preprint for details.
Anyways, we show this leads to good assemblies, especially when similar genomes (within-species diversity) are present. See results for mock ONT R10.4 synthetic community:
3 / N
Anyways, we show this leads to good assemblies, especially when similar genomes (within-species diversity) are present. See results for mock ONT R10.4 synthetic community:
3 / N

September 7, 2025 at 11:35 PM
We do a bunch more assembly graph cleaning using coverages (and even some _theory_!). See preprint for details.
Anyways, we show this leads to good assemblies, especially when similar genomes (within-species diversity) are present. See results for mock ONT R10.4 synthetic community:
3 / N
Anyways, we show this leads to good assemblies, especially when similar genomes (within-species diversity) are present. See results for mock ONT R10.4 synthetic community:
3 / N
We designed a new string graph assembler using polymorphic within-sample k-mers.
We often use SNPs to disentangle similar genomic regions ("inexact repeats", e.g. haplotypes).
But in assembly, no reference exists, so we use a context-free k-mer representation instead ("SNPmers").
2 / N
We often use SNPs to disentangle similar genomic regions ("inexact repeats", e.g. haplotypes).
But in assembly, no reference exists, so we use a context-free k-mer representation instead ("SNPmers").
2 / N

September 7, 2025 at 11:35 PM
We designed a new string graph assembler using polymorphic within-sample k-mers.
We often use SNPs to disentangle similar genomic regions ("inexact repeats", e.g. haplotypes).
But in assembly, no reference exists, so we use a context-free k-mer representation instead ("SNPmers").
2 / N
We often use SNPs to disentangle similar genomic regions ("inexact repeats", e.g. haplotypes).
But in assembly, no reference exists, so we use a context-free k-mer representation instead ("SNPmers").
2 / N
Limitations of myloasm are that it takes slightly more memory and, like other assemblers, can occasionally produce errors.
We try to be upfront about this and discuss it here myloasm-docs.github.io/qc/. We provide additional info for plotting and curation too.
We try to be upfront about this and discuss it here myloasm-docs.github.io/qc/. We provide additional info for plotting and curation too.
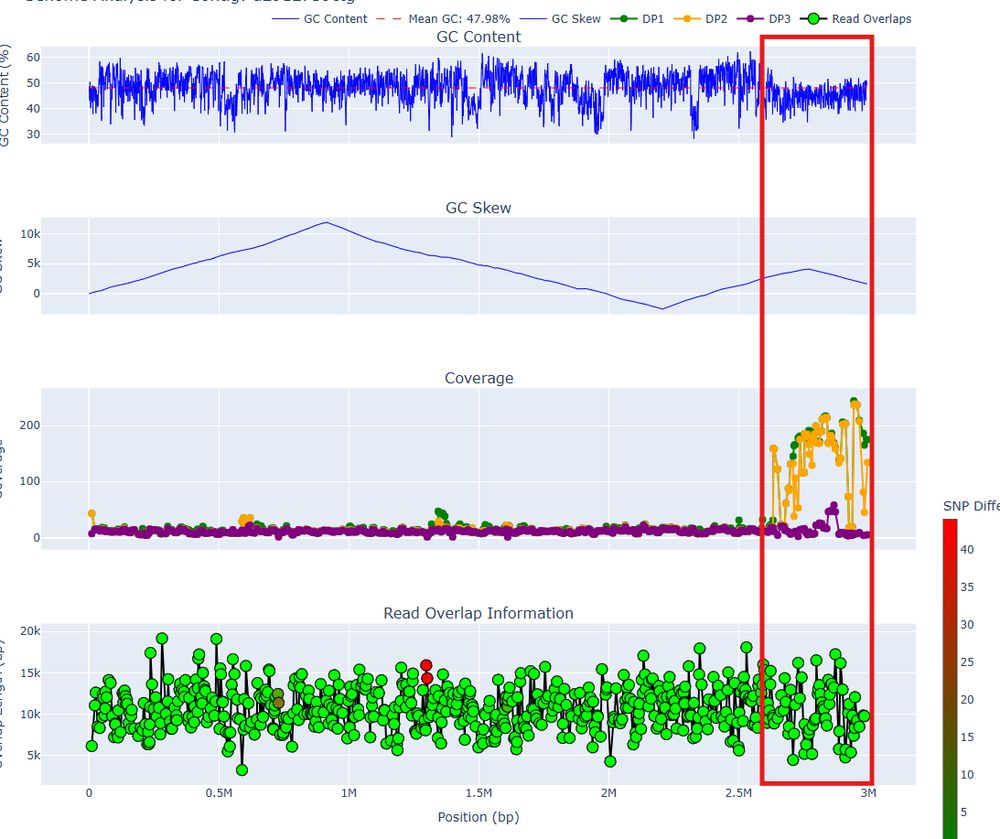
May 28, 2025 at 5:54 PM
Limitations of myloasm are that it takes slightly more memory and, like other assemblers, can occasionally produce errors.
We try to be upfront about this and discuss it here myloasm-docs.github.io/qc/. We provide additional info for plotting and curation too.
We try to be upfront about this and discuss it here myloasm-docs.github.io/qc/. We provide additional info for plotting and curation too.
It seems we can assemble (reasonably simple populations) of co-existing strains with ONT data now.
We assembled 6 single-contig Prevotella copri genomes of > 97% ANI for one metagenome. 4 of them were circular.
(The largest metaFlye P. copri contig was 13.4% complete)
We assembled 6 single-contig Prevotella copri genomes of > 97% ANI for one metagenome. 4 of them were circular.
(The largest metaFlye P. copri contig was 13.4% complete)
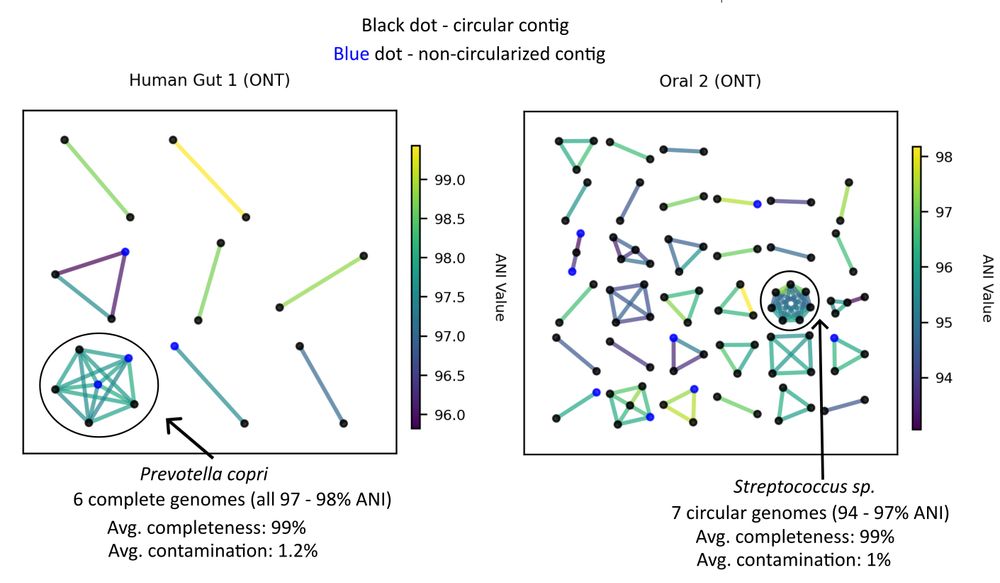
May 28, 2025 at 5:54 PM
It seems we can assemble (reasonably simple populations) of co-existing strains with ONT data now.
We assembled 6 single-contig Prevotella copri genomes of > 97% ANI for one metagenome. 4 of them were circular.
(The largest metaFlye P. copri contig was 13.4% complete)
We assembled 6 single-contig Prevotella copri genomes of > 97% ANI for one metagenome. 4 of them were circular.
(The largest metaFlye P. copri contig was 13.4% complete)
Preprint will come in a couple of months. For a brief algorithmic overview and preliminary results, see myloasm-docs.github.io/results/
The main strength of myloasm: it seems like it can assemble more circularized complete genomes than before, and on diverse metagenomes.
The main strength of myloasm: it seems like it can assemble more circularized complete genomes than before, and on diverse metagenomes.

May 28, 2025 at 5:54 PM
Preprint will come in a couple of months. For a brief algorithmic overview and preliminary results, see myloasm-docs.github.io/results/
The main strength of myloasm: it seems like it can assemble more circularized complete genomes than before, and on diverse metagenomes.
The main strength of myloasm: it seems like it can assemble more circularized complete genomes than before, and on diverse metagenomes.
One aspect devider worked surprisingly well at: studying recombination of antimicrobial resistance (AMR) genes.
We applied devider to a bovine gut metagenome enriched for AMR... and we found lots of interesting mosaic haplotypes. See the recombination blocks in the image (found by GARD).
4/5
We applied devider to a bovine gut metagenome enriched for AMR... and we found lots of interesting mosaic haplotypes. See the recombination blocks in the image (found by GARD).
4/5

December 17, 2024 at 11:32 PM
One aspect devider worked surprisingly well at: studying recombination of antimicrobial resistance (AMR) genes.
We applied devider to a bovine gut metagenome enriched for AMR... and we found lots of interesting mosaic haplotypes. See the recombination blocks in the image (found by GARD).
4/5
We applied devider to a bovine gut metagenome enriched for AMR... and we found lots of interesting mosaic haplotypes. See the recombination blocks in the image (found by GARD).
4/5
Bioinfo friends: devider uses a positional de Bruijn graph approach with sequence-to-graph alignments for path retrieval --- but restricted only to an alphabet of heterozygous SNPs.
Technique inspired by a great paper from Zhou et al. for diploid haplotyping (www.nature.com/articles/s41...)
3/5
Technique inspired by a great paper from Zhou et al. for diploid haplotyping (www.nature.com/articles/s41...)
3/5
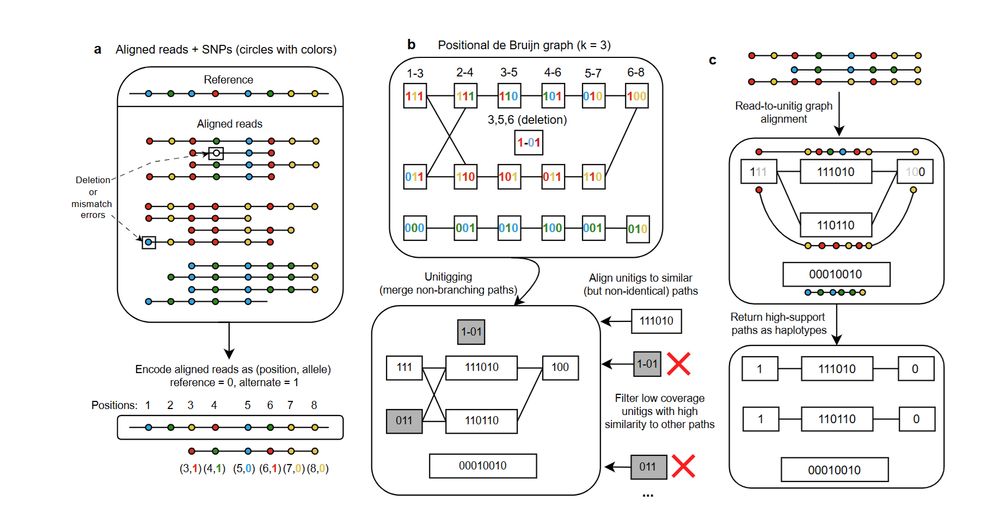
December 17, 2024 at 11:32 PM
Bioinfo friends: devider uses a positional de Bruijn graph approach with sequence-to-graph alignments for path retrieval --- but restricted only to an alphabet of heterozygous SNPs.
Technique inspired by a great paper from Zhou et al. for diploid haplotyping (www.nature.com/articles/s41...)
3/5
Technique inspired by a great paper from Zhou et al. for diploid haplotyping (www.nature.com/articles/s41...)
3/5
In a nutshell, devider can cluster long reads into similar haplotypes (see the images).
Main points:
(1) Works best with sequence length ~= read length
(2) Does not require prior knowledge of # of haplotypes (can be many!)
(3) Extremely fast (> 20,000x coverage is ok!)
(4) Requires reference
2/5
Main points:
(1) Works best with sequence length ~= read length
(2) Does not require prior knowledge of # of haplotypes (can be many!)
(3) Extremely fast (> 20,000x coverage is ok!)
(4) Requires reference
2/5


December 17, 2024 at 11:32 PM
In a nutshell, devider can cluster long reads into similar haplotypes (see the images).
Main points:
(1) Works best with sequence length ~= read length
(2) Does not require prior knowledge of # of haplotypes (can be many!)
(3) Extremely fast (> 20,000x coverage is ok!)
(4) Requires reference
2/5
Main points:
(1) Works best with sequence length ~= read length
(2) Does not require prior knowledge of # of haplotypes (can be many!)
(3) Extremely fast (> 20,000x coverage is ok!)
(4) Requires reference
2/5
We updated our metagenomic profiling + detection tool sylph to v0.5.1 (github.com/bluenote-157...).
Sensitivity for low-abund species is much improved for Illumina now (thanks @fplazaonate for showing issue). bioRxiv updated with a few new benchmarks too.
Sensitivity for low-abund species is much improved for Illumina now (thanks @fplazaonate for showing issue). bioRxiv updated with a few new benchmarks too.

January 26, 2024 at 1:55 AM
We updated our metagenomic profiling + detection tool sylph to v0.5.1 (github.com/bluenote-157...).
Sensitivity for low-abund species is much improved for Illumina now (thanks @fplazaonate for showing issue). bioRxiv updated with a few new benchmarks too.
Sensitivity for low-abund species is much improved for Illumina now (thanks @fplazaonate for showing issue). bioRxiv updated with a few new benchmarks too.