



Erin Young
@erinyoung.bsky.social
Public Health #Bioinformatician. Wants to sequence ALL THE THINGS. Personal account with alternative spellings and grammar structures. She/her
DaisyBlast helps me visualize possible horizontal gene transfer (HGT) events involving plasmids.
November 21, 2025 at 11:41 PM
DaisyBlast helps me visualize possible horizontal gene transfer (HGT) events involving plasmids.
And bioconda
```
conda install -c bioconda daisyblast
```
```
conda install -c bioconda daisyblast
```
November 21, 2025 at 11:41 PM
And bioconda
```
conda install -c bioconda daisyblast
```
```
conda install -c bioconda daisyblast
```

DaisyBlast
A Python tool to find, plot, and export synteny blocks from all-vs-all BLAST.
pypi.org
November 21, 2025 at 11:41 PM
daisyblast sourcecode is up on github github.com/erinyoung/da...

GitHub - erinyoung/daisyblast: Python pipeline for automated synteny block detection and visualization from multiple genomes.
Python pipeline for automated synteny block detection and visualization from multiple genomes. - erinyoung/daisyblast
github.com
November 21, 2025 at 11:41 PM
daisyblast sourcecode is up on github github.com/erinyoung/da...
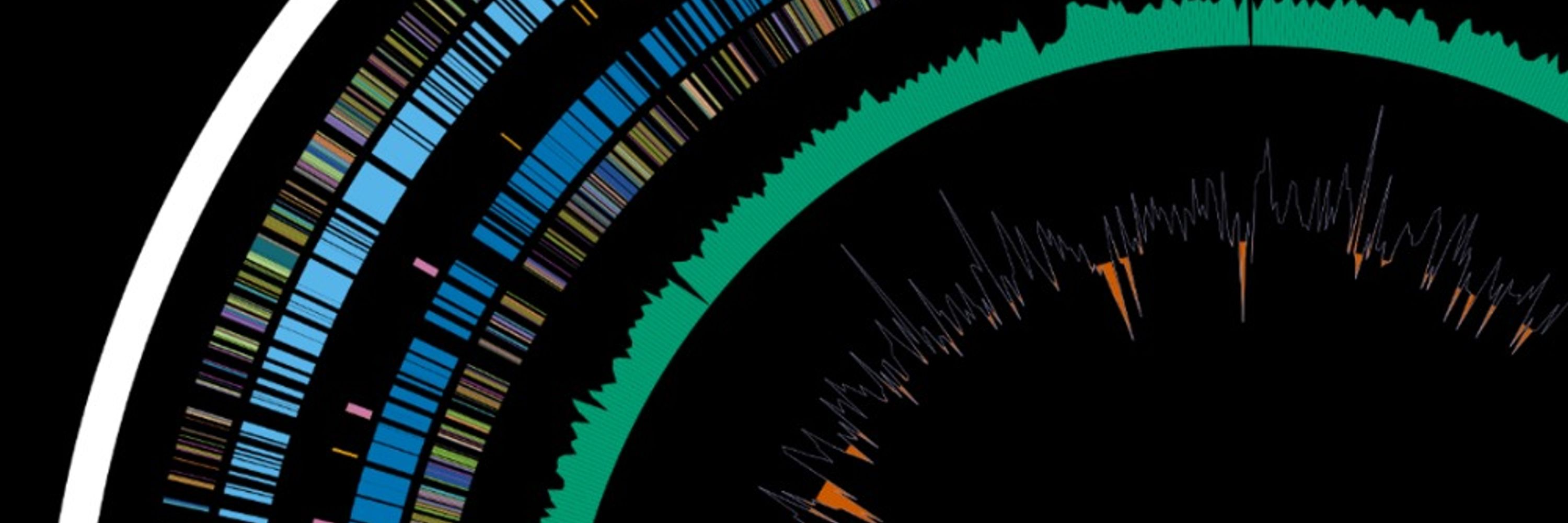
Or little circular figures, also without connecting lines. I've found these impress people, but it is a little harder to identify sections of synteny when the genome is circular.

November 21, 2025 at 11:41 PM
Or little circular figures, also without connecting lines. I've found these impress people, but it is a little harder to identify sections of synteny when the genome is circular.
Or little synteny blocks figures without neighbors (no connecting lines). This is helpful in exploratory analysis where I am initially unsure which sequences are going to be significant.

November 21, 2025 at 11:41 PM
Or little synteny blocks figures without neighbors (no connecting lines). This is helpful in exploratory analysis where I am initially unsure which sequences are going to be significant.
Tumblr: posts a dramatic black-and-white photo of a fin slicing through water with caption “Just circling… thinking about life… and seals…”
November 18, 2025 at 6:01 PM
Tumblr: posts a dramatic black-and-white photo of a fin slicing through water with caption “Just circling… thinking about life… and seals…”
And the cluster link
Isolates SNP Tree Viewer -
Pathogen Detection - NCBI
Isolates SNP Tree Viewer
www.ncbi.nlm.nih.gov
October 17, 2025 at 5:35 PM
And the cluster link
Good idea, but there's more documentation about conda, and a lot of people I work with use the command line less than 5 hours per month.
October 17, 2025 at 3:19 PM
Good idea, but there's more documentation about conda, and a lot of people I work with use the command line less than 5 hours per month.
I personally think it's because each amino acid change gets its own number
October 16, 2025 at 6:27 PM
I personally think it's because each amino acid change gets its own number
It'd really shift my way of thinking if it didn't
September 29, 2025 at 5:05 PM
It'd really shift my way of thinking if it didn't
I think you misunderstand. There is a lot of coverage for these samples. So much so that it is hard to see bubbles or other aberrations.

September 23, 2025 at 8:46 PM
I think you misunderstand. There is a lot of coverage for these samples. So much so that it is hard to see bubbles or other aberrations.
How is this method simpler than what I attempted?
September 23, 2025 at 7:48 PM
How is this method simpler than what I attempted?
All 33 million+ reads were mapped with bbmap (all non-mapped reads were excluded prior to trimming the primers)
September 23, 2025 at 7:38 PM
All 33 million+ reads were mapped with bbmap (all non-mapped reads were excluded prior to trimming the primers)
So, in summary, if high coverage samples aren't getting assigned lineages, I recommend subsampling them or adjusting the `samtools mpileup` command.
September 23, 2025 at 6:01 PM
So, in summary, if high coverage samples aren't getting assigned lineages, I recommend subsampling them or adjusting the `samtools mpileup` command.
I really solved my dilemma (after too many hours trouble shooting it) by adding the `-d 0` flag to `samtools mpileup`, which uses a lot of memory but produced adequate consensus fasta files.
/
/
September 23, 2025 at 6:01 PM
I really solved my dilemma (after too many hours trouble shooting it) by adding the `-d 0` flag to `samtools mpileup`, which uses a lot of memory but produced adequate consensus fasta files.
/
/
Instead it turns out that `bbmap` was allowing for very large insert sizes, some of which spanned the majority of the genome. These, for whatever reason, were given priority for `samtools mpileup` (which gets piped into `ivar consensus`) /
September 23, 2025 at 6:01 PM
Instead it turns out that `bbmap` was allowing for very large insert sizes, some of which spanned the majority of the genome. These, for whatever reason, were given priority for `samtools mpileup` (which gets piped into `ivar consensus`) /
The most irritating part is that each of these 18 would generate a consensus that could be used for determining lineage if I subsampled them. I was worried about contamination, but these had all had human reads removed. /
September 23, 2025 at 6:01 PM
The most irritating part is that each of these 18 would generate a consensus that could be used for determining lineage if I subsampled them. I was worried about contamination, but these had all had human reads removed. /