



Reposted by Brad Chapman

Computational variant effect predictors effectively predict viral evolution – even more so than deep mutational scans (DMS).
Yet, PLM or hybrid approaches (even with data-leakage inflating performance) provide little benefit over the best alignment-based model (EVE).
Yet, PLM or hybrid approaches (even with data-leakage inflating performance) provide little benefit over the best alignment-based model (EVE).

January 13, 2026 at 7:06 PM
Computational variant effect predictors effectively predict viral evolution – even more so than deep mutational scans (DMS).
Yet, PLM or hybrid approaches (even with data-leakage inflating performance) provide little benefit over the best alignment-based model (EVE).
Yet, PLM or hybrid approaches (even with data-leakage inflating performance) provide little benefit over the best alignment-based model (EVE).
Reposted by Brad Chapman

Thrilled to share EDEN — a DNA-scale foundation model built with @basecamp-research.bsky.social + NVIDIA partly validated by our lab @upenn.edu .
Trained on 1M+ new species, it designs novel antibiotics with 97% success. Biodiversity at scale = models that generalize and design.
bit.ly/3NpMR4B
Trained on 1M+ new species, it designs novel antibiotics with 97% success. Biodiversity at scale = models that generalize and design.
bit.ly/3NpMR4B

January 12, 2026 at 6:29 PM
Thrilled to share EDEN — a DNA-scale foundation model built with @basecamp-research.bsky.social + NVIDIA partly validated by our lab @upenn.edu .
Trained on 1M+ new species, it designs novel antibiotics with 97% success. Biodiversity at scale = models that generalize and design.
bit.ly/3NpMR4B
Trained on 1M+ new species, it designs novel antibiotics with 97% success. Biodiversity at scale = models that generalize and design.
bit.ly/3NpMR4B
Reposted by Brad Chapman

🚀 Our new paper on Alignoth just published in Bioinformatics!
Alignoth generates self-contained interactive HTML read alignment plots from BAM files – Rust-based, portable, and ideal for headless workflows.
📄 doi.org/10.1093/bioi...
#bioinformatics #genomics #rust @johanneskoester.bsky.social
Alignoth generates self-contained interactive HTML read alignment plots from BAM files – Rust-based, portable, and ideal for headless workflows.
📄 doi.org/10.1093/bioi...
#bioinformatics #genomics #rust @johanneskoester.bsky.social

January 8, 2026 at 1:57 PM
🚀 Our new paper on Alignoth just published in Bioinformatics!
Alignoth generates self-contained interactive HTML read alignment plots from BAM files – Rust-based, portable, and ideal for headless workflows.
📄 doi.org/10.1093/bioi...
#bioinformatics #genomics #rust @johanneskoester.bsky.social
Alignoth generates self-contained interactive HTML read alignment plots from BAM files – Rust-based, portable, and ideal for headless workflows.
📄 doi.org/10.1093/bioi...
#bioinformatics #genomics #rust @johanneskoester.bsky.social
Reposted by Brad Chapman

Can we design mutations that bias proteins towards desired conformational states?
Today in @science.org, we introduce Conformational Biasing (CB), a simple and scalable computational method that uses contrastive scoring by inverse folding models to identify conformation-biasing mutations.
Today in @science.org, we introduce Conformational Biasing (CB), a simple and scalable computational method that uses contrastive scoring by inverse folding models to identify conformation-biasing mutations.

Computational design of conformation-biasing mutations to alter protein functions
Conformational biasing (CB) is a rapid and streamlined computational method that uses contrastive scoring by inverse folding models to predict protein variants biased toward desired conformational sta...
www.science.org
January 8, 2026 at 7:08 PM
Can we design mutations that bias proteins towards desired conformational states?
Today in @science.org, we introduce Conformational Biasing (CB), a simple and scalable computational method that uses contrastive scoring by inverse folding models to identify conformation-biasing mutations.
Today in @science.org, we introduce Conformational Biasing (CB), a simple and scalable computational method that uses contrastive scoring by inverse folding models to identify conformation-biasing mutations.
Reposted by Brad Chapman
Now published in gigascience: academic.oup.com/gigascience/.... Key messages: SVs are highly enriched in low-complexity/tandem-repeat regions and are harder to call. They behave differently from transposon insertions. Always stratify if you study SVs.
January 6, 2026 at 10:55 PM
Now published in gigascience: academic.oup.com/gigascience/.... Key messages: SVs are highly enriched in low-complexity/tandem-repeat regions and are harder to call. They behave differently from transposon insertions. Always stratify if you study SVs.
Reposted by Brad Chapman
Now published in Algorithms for Molecular Biology: link.springer.com/article/10.1.... Key message: a tiny CNN model with 7k parameters can capture main splice signals across vertebrates+insect and halves the minimap2 & miniprot junction error rate. I always use this new feature now.
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
arxiv.org/abs/2506.12986
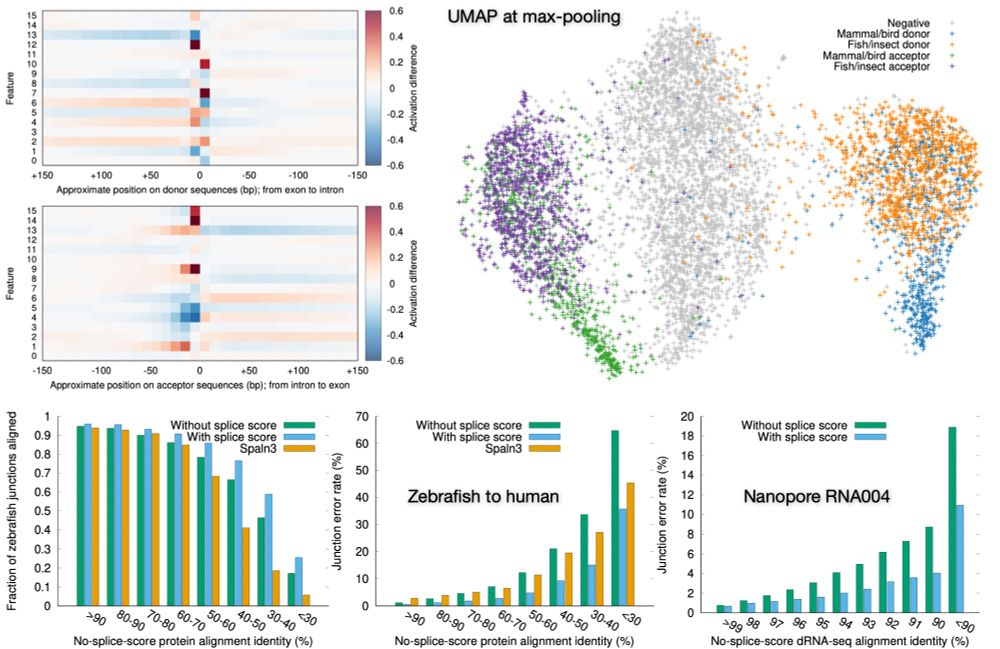
January 6, 2026 at 11:02 PM
Now published in Algorithms for Molecular Biology: link.springer.com/article/10.1.... Key message: a tiny CNN model with 7k parameters can capture main splice signals across vertebrates+insect and halves the minimap2 & miniprot junction error rate. I always use this new feature now.
Reposted by Brad Chapman

New preprint alert: we use sign errors as a test of how well TWAS works.
Very worryingly we find that TWAS gets the sign wrong around 1/3 of the time (compared to 50% for pure guessing). You can read more about our analysis here, and what we think is going on 👇
Very worryingly we find that TWAS gets the sign wrong around 1/3 of the time (compared to 50% for pure guessing). You can read more about our analysis here, and what we think is going on 👇

How well does TWAS estimate a gene’s direction of effect on a trait? We think of this as an important stress-test for the accuracy of TWAS.
In a new pre-print, we find that TWAS gets the sign wrong around 20-30% of the time!
doi.org/10.64898/202...
1/n
In a new pre-print, we find that TWAS gets the sign wrong around 20-30% of the time!
doi.org/10.64898/202...
1/n

High false sign rates in transcriptome-wide association studies
Transcriptome-wide association studies (TWAS) are widely used to identify genes involved in complex traits and to infer the direction of gene effects on traits. However, despite their popularity, it r...
doi.org
January 6, 2026 at 2:48 AM
New preprint alert: we use sign errors as a test of how well TWAS works.
Very worryingly we find that TWAS gets the sign wrong around 1/3 of the time (compared to 50% for pure guessing). You can read more about our analysis here, and what we think is going on 👇
Very worryingly we find that TWAS gets the sign wrong around 1/3 of the time (compared to 50% for pure guessing). You can read more about our analysis here, and what we think is going on 👇
Reposted by Brad Chapman

An interesting and provocative preprint by the Johnson group that challenges the decades old hypothesis that PGR5-cyclic electron flow is required to balance ATP/NADPH requirements for carbon fixation @biorxiv-plants.bsky.social worth a read doi.org/10.1101/2025... #photosynthesis

Disequilibrium between chloroplast proton motive force and ATP levels in Arabidopsis
Current dogma holds that CO2 fixation by photosynthesis requires additional ATP production via PGR5-dependent cyclic electron transfer (PGR5-CET) to augment the NADPH and ATP produced by linear electr...
doi.org
January 2, 2026 at 8:13 PM
An interesting and provocative preprint by the Johnson group that challenges the decades old hypothesis that PGR5-cyclic electron flow is required to balance ATP/NADPH requirements for carbon fixation @biorxiv-plants.bsky.social worth a read doi.org/10.1101/2025... #photosynthesis
Reposted by Brad Chapman

a post about how, with LLMs, I wrote a new set of libraries.
Wrapping htslib is my hello-world but doing it in rust to allow javascript expressions handled by V8 was more substantial project.
The tools and LLMs are now getting quite good:
brentp.github.io/latest/blog/...
let me know what you think
Wrapping htslib is my hello-world but doing it in rust to allow javascript expressions handled by V8 was more substantial project.
The tools and LLMs are now getting quite good:
brentp.github.io/latest/blog/...
let me know what you think
AI Coding - Brent Pedersen
Writing real software with AI coding tools
brentp.github.io
December 30, 2025 at 4:42 PM
a post about how, with LLMs, I wrote a new set of libraries.
Wrapping htslib is my hello-world but doing it in rust to allow javascript expressions handled by V8 was more substantial project.
The tools and LLMs are now getting quite good:
brentp.github.io/latest/blog/...
let me know what you think
Wrapping htslib is my hello-world but doing it in rust to allow javascript expressions handled by V8 was more substantial project.
The tools and LLMs are now getting quite good:
brentp.github.io/latest/blog/...
let me know what you think
Reposted by Brad Chapman

I've been thinking about the "virtual cell" concept and wanted to write up a few thoughts. Specifically on how I think the prior experience in GWAS informs the most likely way these models will be useful.
andrewcarroll.github.io/2025/12/23/t...
andrewcarroll.github.io/2025/12/23/t...
The Virtual Cell Will Be More Like Gwas Than Alphafold
There has been significant discussion recently on the concept of the “virtual cell.” I want to summarize the key concepts regarding what the field wants from a virtual cell and the challenges we face....
andrewcarroll.github.io
December 24, 2025 at 1:52 AM
I've been thinking about the "virtual cell" concept and wanted to write up a few thoughts. Specifically on how I think the prior experience in GWAS informs the most likely way these models will be useful.
andrewcarroll.github.io/2025/12/23/t...
andrewcarroll.github.io/2025/12/23/t...
Reposted by Brad Chapman

Our latest protein family-based GenAI collection of tools and datasets, ProFam, is out now. Everything -- from data, training and inference code, to a 215M llama-based ProFam-1 are fully open sourced.
🧵
🧵

Built by CATH, TÜM and NVIDIA, ProFam-1 is our new open-source protein family language model (pfLM) designed to generate functional protein variants and predict fitness using in-context example sequences.
December 22, 2025 at 2:57 PM
Our latest protein family-based GenAI collection of tools and datasets, ProFam, is out now. Everything -- from data, training and inference code, to a 215M llama-based ProFam-1 are fully open sourced.
🧵
🧵
Reposted by Brad Chapman

Enzyme Engineering Database (EnzEngDB): a platform for sharing and interpreting sequence–function relationships across protein engineering campaigns
@francescazfl.bsky.social @jsunn-y.bsky.social @francesarnold.bsky.social @arianemora.bsky.social
Paper: doi.org/10.1093/nar/...
DB: enzengdb.org
@francescazfl.bsky.social @jsunn-y.bsky.social @francesarnold.bsky.social @arianemora.bsky.social
Paper: doi.org/10.1093/nar/...
DB: enzengdb.org
December 12, 2025 at 8:56 PM
Enzyme Engineering Database (EnzEngDB): a platform for sharing and interpreting sequence–function relationships across protein engineering campaigns
@francescazfl.bsky.social @jsunn-y.bsky.social @francesarnold.bsky.social @arianemora.bsky.social
Paper: doi.org/10.1093/nar/...
DB: enzengdb.org
@francescazfl.bsky.social @jsunn-y.bsky.social @francesarnold.bsky.social @arianemora.bsky.social
Paper: doi.org/10.1093/nar/...
DB: enzengdb.org
Reposted by Brad Chapman

Prediction is overhead when verification is cheap.
We've spent years building a system where verification is cheap -- generationally so.
This changes the logic of discovery in ways that are easy to underestimate. I tried to write a little about what that means: www.jurabio.com/blog/onebill...
We've spent years building a system where verification is cheap -- generationally so.
This changes the logic of discovery in ways that are easy to underestimate. I tried to write a little about what that means: www.jurabio.com/blog/onebill...

One billion simultaneous experiments — JURA Bio, Inc.
For decades, drug discovery has been constrained by a simple fact: experiments are expensive, so you have to guess well. We built a system where you don't have to guess — testing a billion distinct m...
www.jurabio.com
December 12, 2025 at 12:56 PM
Prediction is overhead when verification is cheap.
We've spent years building a system where verification is cheap -- generationally so.
This changes the logic of discovery in ways that are easy to underestimate. I tried to write a little about what that means: www.jurabio.com/blog/onebill...
We've spent years building a system where verification is cheap -- generationally so.
This changes the logic of discovery in ways that are easy to underestimate. I tried to write a little about what that means: www.jurabio.com/blog/onebill...
Reposted by Brad Chapman

1/4 Thrilled to be sharing new work published today in Nature describing the third wave of results from the PGC Cross-Disorder Group. This reflects a massive group effort to examine shared and unique genetic signal across >1 million cases for 14 psychiatric disorders. www.nature.com/articles/s41...

Mapping the genetic landscape across 14 psychiatric disorders - Nature
Genomic analyses applied to 14 childhood- and adult-onset psychiatric disorders identifies five underlying genomic factors that explain the majority of the genetic variance of the individual disorders...
www.nature.com
December 10, 2025 at 4:22 PM
1/4 Thrilled to be sharing new work published today in Nature describing the third wave of results from the PGC Cross-Disorder Group. This reflects a massive group effort to examine shared and unique genetic signal across >1 million cases for 14 psychiatric disorders. www.nature.com/articles/s41...
Reposted by Brad Chapman

Over the past 5+ years I've had the honor of working with @wsdewitt.github.io @victora.bsky.social and many others on a project to "replay" affinity maturation evolution from a fixed starting point.
matsen.group/general/2025...
matsen.group/general/2025...

Replaying evolution to learn about the fitness landscape of affinity maturation
A five year collaboration with the Victora lab is bearing fruit for evolutionary biology.
matsen.group
December 11, 2025 at 5:36 PM
Over the past 5+ years I've had the honor of working with @wsdewitt.github.io @victora.bsky.social and many others on a project to "replay" affinity maturation evolution from a fixed starting point.
matsen.group/general/2025...
matsen.group/general/2025...
Reposted by Brad Chapman

After a huge amount of work w/ @alex-stark.bsky.social's group, a new version of our Ledidi preprint is now out!
In an era of AI-designed proteins, the next leap will be controlling when, where, and how much of these proteins are expressed in living cells.
www.biorxiv.org/content/10.1...
In an era of AI-designed proteins, the next leap will be controlling when, where, and how much of these proteins are expressed in living cells.
www.biorxiv.org/content/10.1...

Programmatic design and editing of cis-regulatory elements
The development of modern genome editing and DNA synthesis has enabled researchers to edit DNA sequences with high precision but has left unsolved the problem of designing these edits. We introduce Le...
www.biorxiv.org
December 10, 2025 at 3:18 PM
After a huge amount of work w/ @alex-stark.bsky.social's group, a new version of our Ledidi preprint is now out!
In an era of AI-designed proteins, the next leap will be controlling when, where, and how much of these proteins are expressed in living cells.
www.biorxiv.org/content/10.1...
In an era of AI-designed proteins, the next leap will be controlling when, where, and how much of these proteins are expressed in living cells.
www.biorxiv.org/content/10.1...
Reposted by Brad Chapman

gffutilsAI: An AI-Agent for Interactive Genomic Feature Exploration in GFF files www.biorxiv.org/content/10.6... 🧬🖥️🧪 github.com/ToyokoLabs/g...

December 6, 2025 at 5:00 PM
gffutilsAI: An AI-Agent for Interactive Genomic Feature Exploration in GFF files www.biorxiv.org/content/10.6... 🧬🖥️🧪 github.com/ToyokoLabs/g...
Reposted by Brad Chapman
579 high-quality human genomes from @humanpangenome.bsky.social, Arab Pangenome and individual papers (CHM13, CN1, KSA001, I002C, YAO and KOREF1). Sequences available in the AGC format (3.7GB) and FM-index in the ropebwt3 format (20.3GB). For details, see github.com/lh3/human-asm

GitHub - lh3/human-asm: A collection of high-quality human genomes
A collection of high-quality human genomes. Contribute to lh3/human-asm development by creating an account on GitHub.
github.com
December 3, 2025 at 3:44 AM
579 high-quality human genomes from @humanpangenome.bsky.social, Arab Pangenome and individual papers (CHM13, CN1, KSA001, I002C, YAO and KOREF1). Sequences available in the AGC format (3.7GB) and FM-index in the ropebwt3 format (20.3GB). For details, see github.com/lh3/human-asm
Reposted by Brad Chapman

We are thrilled to announce the first official release (v0.1.8) of #𝗯𝗲𝗱𝗱𝗲𝗿, the successor to one of our flagship tool, #𝗯𝗲𝗱𝘁𝗼𝗼𝗹𝘀! Based on ideas we conceived of long ago (!), this was achieved thanks to the dedication of Brent Pedersen.
1/n
1/n
Intro to Bedder – The Quinlan Lab
quinlanlab.org
December 2, 2025 at 2:28 AM
Reposted by Brad Chapman

Our new paper is out: we present a quantitative framework to assess how (bio)tech interventions in plant agriculture could meaningfully contribute to climate mitigation. This may be the most important paper I ever publish #SyntheticBiology #ClimateAction #Agriculture #Biochar doi.org/10.1093/plph...

Harnessing plant agriculture to mitigate climate change: A framework to evaluate synthetic biology (and other) interventions
The scale of plant agriculture can be used to mitigate climate change, and meaningful targets (synthetic biology and comparators) can be selected to increa
doi.org
November 27, 2025 at 5:16 AM
Our new paper is out: we present a quantitative framework to assess how (bio)tech interventions in plant agriculture could meaningfully contribute to climate mitigation. This may be the most important paper I ever publish #SyntheticBiology #ClimateAction #Agriculture #Biochar doi.org/10.1093/plph...
Reposted by Brad Chapman

AI model Helixer predicts eukaryotic genes ab initio, directly from a plain text FASTA file.
No RNA-seq.
No protein homology.
No repeats, hints, or curated evidence.
Raw genome → accurate gene models.
Deep learning + HMM, published in @natmethods.nature.com
www.nature.com/articles/s41...
No RNA-seq.
No protein homology.
No repeats, hints, or curated evidence.
Raw genome → accurate gene models.
Deep learning + HMM, published in @natmethods.nature.com
www.nature.com/articles/s41...

Helixer: ab initio prediction of primary eukaryotic gene models combining deep learning and a hidden Markov model - Nature Methods
By leveraging both deep learning and hidden Markov models, Helixer achieves broad taxonomic coverage for ab initio gene annotation of eukaryotic genomes from fungi, plants, vertebrates and invertebrat...
www.nature.com
November 26, 2025 at 6:00 AM
AI model Helixer predicts eukaryotic genes ab initio, directly from a plain text FASTA file.
No RNA-seq.
No protein homology.
No repeats, hints, or curated evidence.
Raw genome → accurate gene models.
Deep learning + HMM, published in @natmethods.nature.com
www.nature.com/articles/s41...
No RNA-seq.
No protein homology.
No repeats, hints, or curated evidence.
Raw genome → accurate gene models.
Deep learning + HMM, published in @natmethods.nature.com
www.nature.com/articles/s41...
Reposted by Brad Chapman

Newest preprint from our lab: a new family of promising synthetic CO2 fixation cycles that may outcompete the Calvin cycle and that Vittorio Rainaldi realized to a great extent in E. coli! Up to 11 heterologous enzymes in a cascade supporting CO2 fixation & growth! www.biorxiv.org/content/10.1...

Modular in vivo engineering of the reductive methylaspartate cycles for synthetic CO2 fixation
Biological carbon fixation is currently limited to seven naturally occurring pathways. Synthetic carbon fixation pathways have the potential to surpass aerobic natural pathways in efficiency, but none...
www.biorxiv.org
November 25, 2025 at 5:54 PM
Newest preprint from our lab: a new family of promising synthetic CO2 fixation cycles that may outcompete the Calvin cycle and that Vittorio Rainaldi realized to a great extent in E. coli! Up to 11 heterologous enzymes in a cascade supporting CO2 fixation & growth! www.biorxiv.org/content/10.1...
Reposted by Brad Chapman

popEVE is out in Nature Genetics! 🎉
We built a proteome-wide model that combines cross-species and human population variation to rank missense variants by disease severity and help diagnose rare genetic disorders.
rdcu.be/eRu7K
We built a proteome-wide model that combines cross-species and human population variation to rank missense variants by disease severity and help diagnose rare genetic disorders.
rdcu.be/eRu7K

Proteome-wide model for human disease genetics
Nature Genetics - popEVE is a proteome-wide deep generative model to identify and predict pathogenicity of missense mutations causing genetic disorders.
rdcu.be
November 24, 2025 at 1:35 PM
popEVE is out in Nature Genetics! 🎉
We built a proteome-wide model that combines cross-species and human population variation to rank missense variants by disease severity and help diagnose rare genetic disorders.
rdcu.be/eRu7K
We built a proteome-wide model that combines cross-species and human population variation to rank missense variants by disease severity and help diagnose rare genetic disorders.
rdcu.be/eRu7K
Reposted by Brad Chapman

Delighted to report that our group's passion project over the last 6+ years to make DNA cloning more accessible, efficient, and scalable using a software-assisted workflow called CloneCoordinate is now out in ACS Synbio!
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...

CloneCoordinate: Open-Source Software for Collaborative DNA Construction
Custom DNA constructs have never been more common or important in the life sciences. Many researchers therefore devote substantial time and effort to molecular cloning, aided by abundant computer-aide...
pubs.acs.org
November 21, 2025 at 7:40 PM
Delighted to report that our group's passion project over the last 6+ years to make DNA cloning more accessible, efficient, and scalable using a software-assisted workflow called CloneCoordinate is now out in ACS Synbio!
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...
Reposted by Brad Chapman
The last five months with Claude Code have completely changed how we work.
matsen.group/agentic.html details:
• How agents work (& why it matters)
• Git Flow with agents
• Using agents for science
• The human-agent interface
Questions? What has your experience been?
matsen.group/agentic.html details:
• How agents work (& why it matters)
• Git Flow with agents
• Using agents for science
• The human-agent interface
Questions? What has your experience been?

Agentic Coding For Scientists
A four-part series on using coding agents like Claude Code for scientific programming, covering fundamentals, workflows, best practices, and the human side of AI-assisted development.
matsen.group
November 13, 2025 at 5:08 PM
The last five months with Claude Code have completely changed how we work.
matsen.group/agentic.html details:
• How agents work (& why it matters)
• Git Flow with agents
• Using agents for science
• The human-agent interface
Questions? What has your experience been?
matsen.group/agentic.html details:
• How agents work (& why it matters)
• Git Flow with agents
• Using agents for science
• The human-agent interface
Questions? What has your experience been?