



Rick Beeloo
@rickbitloo.bsky.social
PhD candidate Utrecht University
Some bio and coding stuff
Some bio and coding stuff
Reposted by Rick Beeloo

If you ever need to fuzzy search some DNA, sassy is your tool.
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
December 10, 2025 at 3:50 PM
If you ever need to fuzzy search some DNA, sassy is your tool.
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
Reposted by Rick Beeloo
Following ish's `filter` and bqtools' `grep`, Sassy now also has initial support for grep and filter!
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.

October 30, 2025 at 11:46 PM
Following ish's `filter` and bqtools' `grep`, Sassy now also has initial support for grep and filter!
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.
Reposted by Rick Beeloo
Really exciting that the preprint on Barbell, a new demultiplexer, is finally out!
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
Around 10% of your Nanopore reads (SQK-RBK114) are incorrectly trimmed. Here is why, and how our new tool Barbell solves it:
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
October 23, 2025 at 9:28 PM
Really exciting that the preprint on Barbell, a new demultiplexer, is finally out!
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
Reposted by Rick Beeloo

Barbell Resolves Demultiplexing and Trimming Issues in Nanopore Data https://www.biorxiv.org/content/10.1101/2025.10.22.683865v1
October 23, 2025 at 7:47 PM
Barbell Resolves Demultiplexing and Trimming Issues in Nanopore Data https://www.biorxiv.org/content/10.1101/2025.10.22.683865v1
Around 10% of your Nanopore reads (SQK-RBK114) are incorrectly trimmed. Here is why, and how our new tool Barbell solves it:
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
October 23, 2025 at 8:16 PM
Around 10% of your Nanopore reads (SQK-RBK114) are incorrectly trimmed. Here is why, and how our new tool Barbell solves it:
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
Reposted by Rick Beeloo
Now also on biorxiv :)

Sassy: Searching Short DNA Strings in the 2020s https://www.biorxiv.org/content/10.1101/2025.07.22.666207v1
July 26, 2025 at 7:15 PM
Now also on biorxiv :)
Reposted by Rick Beeloo
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
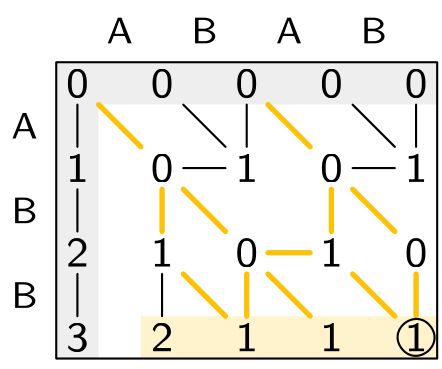
July 18, 2025 at 8:20 PM
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...