




Thilo Muth
@drmuth.bsky.social
Group Leader and Adjunct Professor 🧑🏫 | Public Health | Data Engineering & Visualization | 🧬 Bioinformatics & 📈 Mass Spec Enthusiast | 📍 Berlin | Passionate About Collaboration and Open Science 🔬✨
Reposted by Thilo Muth

Researchers have performed a large-scale genetic screen to uncover the hidden roles of #microproteins. One of the discovered microproteins, named PIPPI, was found to protect cells from stress in the endoplasmic reticulum. Published in @narjournal.bsky.social 🧪 #proteomics news.ki.se/new-micropro...

New microprotein can help cancer cells overcome stress
In a new study published in the journal Nucleic Acid Research, a research team at Karolinska Institutet has performed a large-scale genetic screen to uncover the hidden roles of tiny proteins, so-call...
news.ki.se
November 27, 2025 at 7:55 AM
Researchers have performed a large-scale genetic screen to uncover the hidden roles of #microproteins. One of the discovered microproteins, named PIPPI, was found to protect cells from stress in the endoplasmic reticulum. Published in @narjournal.bsky.social 🧪 #proteomics news.ki.se/new-micropro...
Reposted by Thilo Muth

Cannot hide my joy, relief, excitement being able to share the outcome of persevering (to say the least) collaborative work. So... the MS glyco(proteomics) guidelines are published!! www.mcponline.org/article/S153...
Many thanks to @beilstein-institut.bsky.social @glycosmos.bsky.social @sib.swiss
Many thanks to @beilstein-institut.bsky.social @glycosmos.bsky.social @sib.swiss

Update and new implementation of the MIRAGE reporting guidelines for mass spectrometry experiments in glycoscience
The MIRAGE (Minimum Information Required for A Glycomics Experiment) guidelines for
mass spectrometry (MS) data were initially developed to standardize the reporting
of instrumentation, data acquisiti...
www.mcponline.org
November 25, 2025 at 9:34 AM
Cannot hide my joy, relief, excitement being able to share the outcome of persevering (to say the least) collaborative work. So... the MS glyco(proteomics) guidelines are published!! www.mcponline.org/article/S153...
Many thanks to @beilstein-institut.bsky.social @glycosmos.bsky.social @sib.swiss
Many thanks to @beilstein-institut.bsky.social @glycosmos.bsky.social @sib.swiss
Reposted by Thilo Muth

It’s the first time that proteomics has been used to analyze Renaissance recipes. The project demonstrates an innovative way to study medicine and the circulation and use of medical recipes from the past. cen.acs.org/analytical-c... #chemsky 🧪

Proteins plucked from the pages of Renaissance recipes
Analyses of German medical manuals from 1531 reveal what ingredients those who handled the books used
cen.acs.org
November 27, 2025 at 6:16 PM
It’s the first time that proteomics has been used to analyze Renaissance recipes. The project demonstrates an innovative way to study medicine and the circulation and use of medical recipes from the past. cen.acs.org/analytical-c... #chemsky 🧪
Reposted by Thilo Muth

This project started 5 years ago. It led us to add isotope-labeling support to #FragPipe/#IonQuant. Since then, the tools have grown so much and are now widely used in #Chemoproteomics.
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social

How can we study target engagement and selectivity of covalent inhibitors? Which electrophilic probes are best suited to study a certain amino acid?
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...

Profiling the proteome-wide selectivity of diverse electrophiles - Nature Chemistry
Covalent inhibitors are powerful entities in drug discovery. Now the amino acid selectivity and reactivity of a diverse electrophile library have been assessed proteome-wide using an unbiased workflow...
www.nature.com
October 30, 2025 at 2:15 PM
This project started 5 years ago. It led us to add isotope-labeling support to #FragPipe/#IonQuant. Since then, the tools have grown so much and are now widely used in #Chemoproteomics.
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social
ProteomeXchange consortium in 2026: making proteomics data FAIR url: academic.oup.com/nar/article/...

The ProteomeXchange consortium in 2026: making proteomics data FAIR
Abstract. The ProteomeXchange consortium of proteomics resources (http://www.proteomexchange.org) was established to standardize open data practices in the
academic.oup.com
November 9, 2025 at 7:49 PM
ProteomeXchange consortium in 2026: making proteomics data FAIR url: academic.oup.com/nar/article/...
Reposted by Thilo Muth

On the event of James Watson's death, I highly recommend this 2023 commentary from @matthewcobb.bsky.social and Nathaniel Comfort with crucial new insights into the discovery of the double helix. (And also check out Cobb's brand new biography of Francis Crick) www.nature.com/articles/d41...

What Rosalind Franklin truly contributed to the discovery of DNA’s structure
Franklin was no victim in how the DNA double helix was solved. An overlooked letter and an unpublished news article, both written in 1953, reveal that she was an equal player.
www.nature.com
November 7, 2025 at 9:25 PM
On the event of James Watson's death, I highly recommend this 2023 commentary from @matthewcobb.bsky.social and Nathaniel Comfort with crucial new insights into the discovery of the double helix. (And also check out Cobb's brand new biography of Francis Crick) www.nature.com/articles/d41...
Reposted by Thilo Muth

I think about this post every day 🧪

November 7, 2025 at 12:15 PM
I think about this post every day 🧪
Reposted by Thilo Muth

The reaction to this is fascinating. It's like a microcosm of all discussions around AI: lots of enthusiasm, lots of loathing (much of it reflexive), and some wise 'let's maybe try it and see' responses.
Excited to launch an openRxiv partnership with the scientist-run AI review service qed (@qedscience.bsky.social), the brainchild of @odedrechavi.bsky.social 1/n
openrxiv.org/enabling-rev...
openrxiv.org/enabling-rev...

Enabling options for review: from training and transparency to author-centered AI tools - openRxiv
Peer review is widely viewed as a critical aspect of biomedical communication. Ideally, it provides authors with feedback so they can improve manuscripts and gives readers, particularly nonspecialists...
openrxiv.org
November 7, 2025 at 1:43 PM
The reaction to this is fascinating. It's like a microcosm of all discussions around AI: lots of enthusiasm, lots of loathing (much of it reflexive), and some wise 'let's maybe try it and see' responses.
Reposted by Thilo Muth

Finally, the R package we needed, but don't deserve. I can already hear @benneely.com cackling from across the country.
Thanks to @willfondrie.com for alerting me to this amazing repo.
Thanks to @willfondrie.com for alerting me to this amazing repo.

Do you teach #rstats? Do your students complain about how lame and old-fashioned dplyr is? Don't worry: I have the solution for you: github.com/hadley/genzp....
genzplyr is dplyr, but bussin fr fr no cap.
genzplyr is dplyr, but bussin fr fr no cap.

GitHub - hadley/genzplyr: dplyr but make it bussin fr fr no cap
dplyr but make it bussin fr fr no cap. Contribute to hadley/genzplyr development by creating an account on GitHub.
github.com
November 7, 2025 at 6:38 PM
Finally, the R package we needed, but don't deserve. I can already hear @benneely.com cackling from across the country.
Thanks to @willfondrie.com for alerting me to this amazing repo.
Thanks to @willfondrie.com for alerting me to this amazing repo.
Reposted by Thilo Muth

A previously uncharacterized microbial enzyme is responsible for the production of molecular hydrogen in the gut, which drives the growth of other bacteria and has implications for human health, according to a paper in Nature Microbiology. go.nature.com/4oKOveG #microbiome #medsky 🧪

November 5, 2025 at 2:08 PM
A previously uncharacterized microbial enzyme is responsible for the production of molecular hydrogen in the gut, which drives the growth of other bacteria and has implications for human health, according to a paper in Nature Microbiology. go.nature.com/4oKOveG #microbiome #medsky 🧪
Reposted by Thilo Muth

🧬 Thrilled to share our latest paper in @natmicrobiol.nature.com 📄
A collaboration to give the Flaviviridae (home to Zika, Dengue & HCV) a much-needed taxonomic re-think.
Our at-scale AI structure prediction gave a complementary perspective on viral evolution.
www.nature.com/articles/s41...
A collaboration to give the Flaviviridae (home to Zika, Dengue & HCV) a much-needed taxonomic re-think.
Our at-scale AI structure prediction gave a complementary perspective on viral evolution.
www.nature.com/articles/s41...

Taxonomic expansion and reorganization of Flaviviridae - Nature Microbiology
Analysis of RNA polymerase hallmark gene phylogenies supported by protein structure relationships of flaviviruses and ‘flavi-like’ viruses underpins the taxonomic expansion and reorganization of Flavi...
www.nature.com
November 6, 2025 at 12:52 PM
🧬 Thrilled to share our latest paper in @natmicrobiol.nature.com 📄
A collaboration to give the Flaviviridae (home to Zika, Dengue & HCV) a much-needed taxonomic re-think.
Our at-scale AI structure prediction gave a complementary perspective on viral evolution.
www.nature.com/articles/s41...
A collaboration to give the Flaviviridae (home to Zika, Dengue & HCV) a much-needed taxonomic re-think.
Our at-scale AI structure prediction gave a complementary perspective on viral evolution.
www.nature.com/articles/s41...
Reposted by Thilo Muth
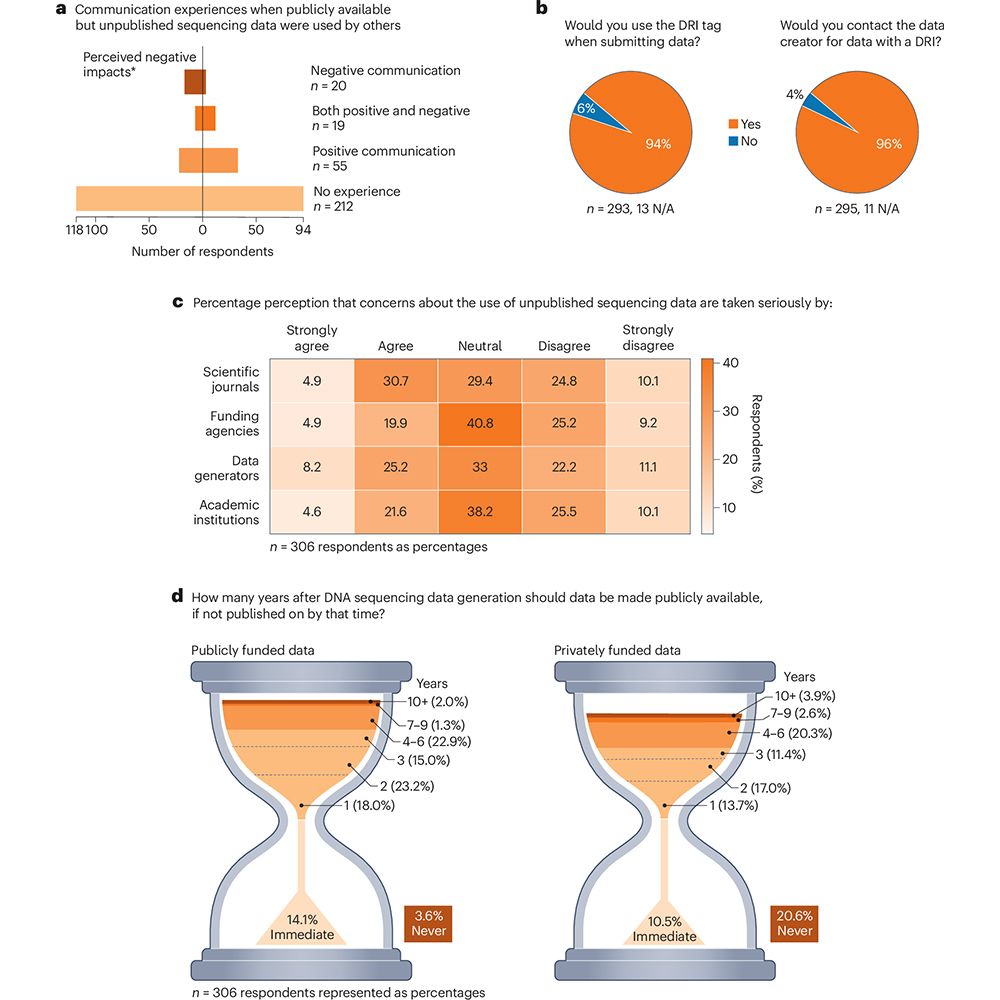
In a Consensus Statement in Nature Microbiology, a consortium of #microbiome scientists discusses current sequencing data sharing policies and proposes the use of a Data Reuse Information tag to promote equitable and collaborative data sharing. go.nature.com/4o1Gl1f 🧪
September 30, 2025 at 1:15 PM
In a Consensus Statement in Nature Microbiology, a consortium of #microbiome scientists discusses current sequencing data sharing policies and proposes the use of a Data Reuse Information tag to promote equitable and collaborative data sharing. go.nature.com/4o1Gl1f 🧪
Reposted by Thilo Muth

Our latest paper introduces new methods for improving peptide retention time predictions in proteomics, incorporating chemical structure information to better handle unseen modifications.
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning

iDeepLC: chemical structure information yields improved retention time prediction of peptides with unseen modifications
Deep learning has notably advanced the field of liquid chromatography–mass spectrometry-based proteomics. Accurate prediction of peptide retention times significantly enhances our ability to match LC-...
doi.org
November 4, 2025 at 8:53 PM
Our latest paper introduces new methods for improving peptide retention time predictions in proteomics, incorporating chemical structure information to better handle unseen modifications.
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning
Carafe - spectral library prediction for DIA proteomics
www.nature.com/articles/s41...
www.nature.com/articles/s41...

Carafe enables high quality in silico spectral library generation for data-independent acquisition proteomics - Nature Communications
Accurate spectral libraries are essential for analyzing data-independent acquisition (DIA) proteomics data. Here, the authors present Carafe, which trains on DIA data to build experiment-specific spec...
www.nature.com
November 7, 2025 at 7:29 PM
Carafe - spectral library prediction for DIA proteomics
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Reposted by Thilo Muth

If you are interested in mRNA vaccines against bacteria, take a look at our recent review. It was a pleasure to write this together with all co-authors! rdcu.be/eyjBq
www.nature.com/articles/s41...
www.nature.com/articles/s41...
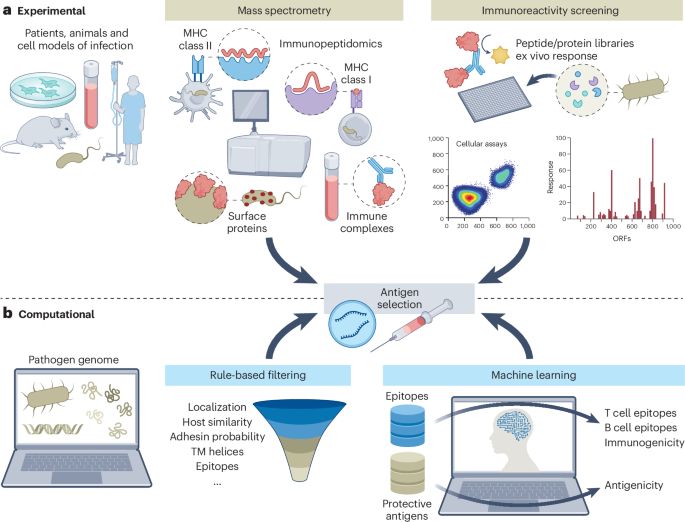
Challenges and opportunities in mRNA vaccine development against bacteria - Nature Microbiology
This Review reflects on the major challenges in bacterial mRNA vaccine design, provides strategies for tailoring mRNA construct design to promote humoral or cellular immunity, and provides an overview...
www.nature.com
August 1, 2025 at 7:11 PM
If you are interested in mRNA vaccines against bacteria, take a look at our recent review. It was a pleasure to write this together with all co-authors! rdcu.be/eyjBq
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Reposted by Thilo Muth

“We fundamentally believe that publishing less – but better – is essential for the health of the entire research system worldwide,” the authors of the report state.

Less is more: academic publishing needs ‘radical change,’ Cambridge press report concludes
Academic publishing needs “renewed focus and collective action” to embrace new approaches and ensure the future of the industry, concludes a report from Cambridge University Press, released last we…
retractionwatch.com
October 21, 2025 at 7:11 PM
“We fundamentally believe that publishing less – but better – is essential for the health of the entire research system worldwide,” the authors of the report state.
Reposted by Thilo Muth

From Identification to Insight: Making Full Use of the Diagnostic Potential of MS/MS Proteotyping in Clinical Microbiology Using Efficient Bioinformatics pubs.acs.org/doi/10....
---
#proteomics #prot-paper
---
#proteomics #prot-paper

October 29, 2025 at 4:20 PM
From Identification to Insight: Making Full Use of the Diagnostic Potential of MS/MS Proteotyping in Clinical Microbiology Using Efficient Bioinformatics pubs.acs.org/doi/10....
---
#proteomics #prot-paper
---
#proteomics #prot-paper
Reposted by Thilo Muth

Good news! The #USHUPO2026 abstract submission deadline has been extended through November 19.
Take advantage of this extra time to share your latest proteomics discoveries.
Submit now: www.ushupoconference.org
Take advantage of this extra time to share your latest proteomics discoveries.
Submit now: www.ushupoconference.org

October 30, 2025 at 8:31 PM
Good news! The #USHUPO2026 abstract submission deadline has been extended through November 19.
Take advantage of this extra time to share your latest proteomics discoveries.
Submit now: www.ushupoconference.org
Take advantage of this extra time to share your latest proteomics discoveries.
Submit now: www.ushupoconference.org
Reposted by Thilo Muth
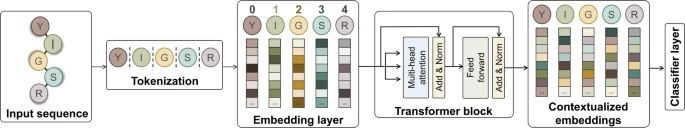
Prediction of peptide cleavage sites using protein language models and graph neural networks www.nature.com/artic...
---
#proteomics #prot-paper
---
#proteomics #prot-paper
October 31, 2025 at 10:00 AM
Prediction of peptide cleavage sites using protein language models and graph neural networks www.nature.com/artic...
---
#proteomics #prot-paper
---
#proteomics #prot-paper
Reposted by Thilo Muth
A shrinkage-based statistical method for testing group mean differences in quantitative bottom-up proteomics - BMC Bioinformatics bmcbioinformatics.bi...
---
#proteomics #prot-paper
---
#proteomics #prot-paper

October 31, 2025 at 1:20 PM
A shrinkage-based statistical method for testing group mean differences in quantitative bottom-up proteomics - BMC Bioinformatics bmcbioinformatics.bi...
---
#proteomics #prot-paper
---
#proteomics #prot-paper
DicePlot: A package for high dimensional categorical data visualization url: academic.oup.com/bioinformati...

DicePlot: A package for high dimensional categorical data visualization
AbstractSummary. Visualization of multidimensional, categorical data is a common challenge across scientific domains and, in particular, the life sciences.
academic.oup.com
November 1, 2025 at 8:22 PM
DicePlot: A package for high dimensional categorical data visualization url: academic.oup.com/bioinformati...
A Phosphoproteomic Analysis of Mycobacterial PknG-Mediated Host Immune Evasion | Journal of Proteome Research pubs.acs.org/doi/10.1021/...

A Phosphoproteomic Analysis of Mycobacterial PknG-Mediated Host Immune Evasion
Pathogenic mycobacteria, such as Mycobacterium tuberculosis, modulate the host immune system to evade clearance and promote long-term persistence, leading to disease progression or latent infection. U...
pubs.acs.org
November 1, 2025 at 8:15 PM
A Phosphoproteomic Analysis of Mycobacterial PknG-Mediated Host Immune Evasion | Journal of Proteome Research pubs.acs.org/doi/10.1021/...
Evaluation of a Prototype Orbitrap Astral Zoom Mass Spectrometer for Quantitative Proteomics─Beyond Identification Lists | Journal of Proteome Research pubs.acs.org/doi/10.1021/...

Evaluation of a Prototype Orbitrap Astral Zoom Mass Spectrometer for Quantitative Proteomics─Beyond Identification Lists
Mass spectrometry instrumentation continues to evolve rapidly, yet quantifying these advances beyond conventional peptide and protein detections remains challenging. Here, we evaluate a modified Orbit...
pubs.acs.org
November 1, 2025 at 8:14 PM
Evaluation of a Prototype Orbitrap Astral Zoom Mass Spectrometer for Quantitative Proteomics─Beyond Identification Lists | Journal of Proteome Research pubs.acs.org/doi/10.1021/...
Data-Independent Immunopeptidomics Discovery of Low-Abundant Bacterial Epitopes | Journal of Proteome Research pubs.acs.org/doi/10.1021/...

Data-Independent Immunopeptidomics Discovery of Low-Abundant Bacterial Epitopes
Mass spectrometry-based immunopeptidomics is a powerful approach to uncover peptides presented by human leukocyte antigen (HLA) molecules that can guide vaccine design and immunotherapies. While data-...
pubs.acs.org
November 1, 2025 at 8:11 PM
Data-Independent Immunopeptidomics Discovery of Low-Abundant Bacterial Epitopes | Journal of Proteome Research pubs.acs.org/doi/10.1021/...
Reposted by Thilo Muth

MRM generation using adaptive modeling of tandem MS data. A simple concept that allows for MRM analysis without the need for standards.
• Especially useful for preclinical metabolites, that have no standards.
Analytical Chemistry (open access article)
pubs.acs.org/doi/10.1021/...
• Especially useful for preclinical metabolites, that have no standards.
Analytical Chemistry (open access article)
pubs.acs.org/doi/10.1021/...
October 30, 2025 at 1:35 PM
MRM generation using adaptive modeling of tandem MS data. A simple concept that allows for MRM analysis without the need for standards.
• Especially useful for preclinical metabolites, that have no standards.
Analytical Chemistry (open access article)
pubs.acs.org/doi/10.1021/...
• Especially useful for preclinical metabolites, that have no standards.
Analytical Chemistry (open access article)
pubs.acs.org/doi/10.1021/...