


Bioinformatics Advances
@bioinfoadv.bsky.social
A fully open access, peer-reviewed journal published jointly by Oxford University Press and the International Society for Computational Biology.
🧫 Explore the latest from Bioinformatics Advances: “BacExplorer: An integrated platform for de novo bacterial genome annotation.”
Full article available: https://doi.org/10.1093/bioadv/vbaf281
Full article available: https://doi.org/10.1093/bioadv/vbaf281

November 28, 2025 at 10:02 AM
🧫 Explore the latest from Bioinformatics Advances: “BacExplorer: An integrated platform for de novo bacterial genome annotation.”
Full article available: https://doi.org/10.1093/bioadv/vbaf281
Full article available: https://doi.org/10.1093/bioadv/vbaf281
🧪 Now published in Bioinformatics Advances: “Are the tools fit for purpose? Network inference algorithms evaluated on a simulated lipidomics network.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf286
Authors include: @rnaprof.bsky.social
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf286
Authors include: @rnaprof.bsky.social

November 27, 2025 at 10:01 AM
🧪 Now published in Bioinformatics Advances: “Are the tools fit for purpose? Network inference algorithms evaluated on a simulated lipidomics network.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf286
Authors include: @rnaprof.bsky.social
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf286
Authors include: @rnaprof.bsky.social
🧫 Just out in Bioinformatics Advances: “GlobDB: A comprehensive species-dereplicated microbial genome resource.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf280
Explore the full study: https://doi.org/10.1093/bioadv/vbaf280

November 26, 2025 at 10:01 AM
🧫 Just out in Bioinformatics Advances: “GlobDB: A comprehensive species-dereplicated microbial genome resource.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf280
Explore the full study: https://doi.org/10.1093/bioadv/vbaf280
🌍 Now published in Bioinformatics Advances: “ntRoot: Computational inference of human ancestry at scale from genomic data.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf287
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf287

November 25, 2025 at 10:01 AM
🌍 Now published in Bioinformatics Advances: “ntRoot: Computational inference of human ancestry at scale from genomic data.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf287
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf287
🦠 Now published in Bioinformatics Advances: “BEREN: A bioinformatic tool for recovering giant viruses, polinton-like viruses, and virophages in metagenomic data.”
Full article available: https://doi.org/10.1093/bioadv/vbaf284
Full article available: https://doi.org/10.1093/bioadv/vbaf284

November 24, 2025 at 10:01 AM
🦠 Now published in Bioinformatics Advances: “BEREN: A bioinformatic tool for recovering giant viruses, polinton-like viruses, and virophages in metagenomic data.”
Full article available: https://doi.org/10.1093/bioadv/vbaf284
Full article available: https://doi.org/10.1093/bioadv/vbaf284
🧠 Now published in Bioinformatics Advances: “Matrix-based vector representations in neural networks for classifying molecular biology data.”
Read the full paper here:
Read the full paper here:
doi.org
November 21, 2025 at 10:01 AM
🧠 Now published in Bioinformatics Advances: “Matrix-based vector representations in neural networks for classifying molecular biology data.”
Read the full paper here:
Read the full paper here:
🧩 Explore the latest from Bioinformatics Advances: “ProCaliper: Functional and structural analysis, visualization, and annotation of proteins.”
Full article available: https://doi.org/10.1093/bioadv/vbaf275
Authors include: @genomesevolve.bsky.social, @songfeng.bsky.social
Full article available: https://doi.org/10.1093/bioadv/vbaf275
Authors include: @genomesevolve.bsky.social, @songfeng.bsky.social

November 20, 2025 at 10:01 AM
🧩 Explore the latest from Bioinformatics Advances: “ProCaliper: Functional and structural analysis, visualization, and annotation of proteins.”
Full article available: https://doi.org/10.1093/bioadv/vbaf275
Authors include: @genomesevolve.bsky.social, @songfeng.bsky.social
Full article available: https://doi.org/10.1093/bioadv/vbaf275
Authors include: @genomesevolve.bsky.social, @songfeng.bsky.social
🧬 Just out in Bioinformatics Advances: “pyALRA: Python implementation of low-rank zero-preserving approximation of single cell RNA-seq.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf279
Explore the full study: https://doi.org/10.1093/bioadv/vbaf279

November 19, 2025 at 10:02 AM
🧬 Just out in Bioinformatics Advances: “pyALRA: Python implementation of low-rank zero-preserving approximation of single cell RNA-seq.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf279
Explore the full study: https://doi.org/10.1093/bioadv/vbaf279
💊 Now published in Bioinformatics Advances: “WebCMap: An R package for high-throughput connectivity analysis within the CMap framework.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf278
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf278

November 18, 2025 at 10:02 AM
💊 Now published in Bioinformatics Advances: “WebCMap: An R package for high-throughput connectivity analysis within the CMap framework.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf278
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf278
🧫 Just out in Bioinformatics Advances: “zAMP and zAMPExplorer: Reproducible scalable amplicon-based metagenomics analysis and visualization.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf255
Explore the full study: https://doi.org/10.1093/bioadv/vbaf255

November 17, 2025 at 10:02 AM
🧫 Just out in Bioinformatics Advances: “zAMP and zAMPExplorer: Reproducible scalable amplicon-based metagenomics analysis and visualization.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf255
Explore the full study: https://doi.org/10.1093/bioadv/vbaf255
🩸 Explore the latest from Bioinformatics Advances: “An interactive mindmap of blood film images for automated malaria diagnosis: a comprehensive metadata repository of available datasets.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf276
Explore the full study: https://doi.org/10.1093/bioadv/vbaf276

November 14, 2025 at 10:01 AM
🩸 Explore the latest from Bioinformatics Advances: “An interactive mindmap of blood film images for automated malaria diagnosis: a comprehensive metadata repository of available datasets.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf276
Explore the full study: https://doi.org/10.1093/bioadv/vbaf276
🧬 Just out in Bioinformatics Advances: “CLUES2 Companion: Computational pipelines to estimate, visualize, and date selection on multi-locus sites.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf277
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf277

November 13, 2025 at 10:02 AM
🧬 Just out in Bioinformatics Advances: “CLUES2 Companion: Computational pipelines to estimate, visualize, and date selection on multi-locus sites.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf277
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf277
🧫 Now published in Bioinformatics Advances: “scExplorer: A comprehensive web server for single-cell RNA sequencing data analysis.”
Full article available: https://doi.org/10.1093/bioadv/vbaf273
Authors include: @fvillanelo.bsky.social
Full article available: https://doi.org/10.1093/bioadv/vbaf273
Authors include: @fvillanelo.bsky.social
doi.org
November 12, 2025 at 10:01 AM
🧫 Now published in Bioinformatics Advances: “scExplorer: A comprehensive web server for single-cell RNA sequencing data analysis.”
Full article available: https://doi.org/10.1093/bioadv/vbaf273
Authors include: @fvillanelo.bsky.social
Full article available: https://doi.org/10.1093/bioadv/vbaf273
Authors include: @fvillanelo.bsky.social
🧬 Now published in Bioinformatics Advances: “LSTM-based deep learning model for the discovery of antimicrobial peptides targeting Mycobacterium tuberculosis.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf274
Authors include: @taanec.bsky.social
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf274
Authors include: @taanec.bsky.social

November 11, 2025 at 10:01 AM
🧬 Now published in Bioinformatics Advances: “LSTM-based deep learning model for the discovery of antimicrobial peptides targeting Mycobacterium tuberculosis.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf274
Authors include: @taanec.bsky.social
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf274
Authors include: @taanec.bsky.social
🧠 Just out in Bioinformatics Advances: “Deep learning for regulatory genomics: A survey of models, challenges, and applications.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf271
Explore the full study: https://doi.org/10.1093/bioadv/vbaf271

November 10, 2025 at 10:02 AM
🧠 Just out in Bioinformatics Advances: “Deep learning for regulatory genomics: A survey of models, challenges, and applications.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf271
Explore the full study: https://doi.org/10.1093/bioadv/vbaf271
🧩 Explore the latest from Bioinformatics Advances: “CoRTE: A web-service for constructing temporal networks from genotype-tissue expression data.”
Full article available: https://doi.org/10.1093/bioadv/vbaf272
Full article available: https://doi.org/10.1093/bioadv/vbaf272

November 7, 2025 at 10:02 AM
🧩 Explore the latest from Bioinformatics Advances: “CoRTE: A web-service for constructing temporal networks from genotype-tissue expression data.”
Full article available: https://doi.org/10.1093/bioadv/vbaf272
Full article available: https://doi.org/10.1093/bioadv/vbaf272
🧠 Now published in Bioinformatics Advances: “PSO-FeatureFusion: A general framework for fusing heterogeneous features via particle swarm optimization.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf263
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf263

November 6, 2025 at 10:02 AM
🧠 Now published in Bioinformatics Advances: “PSO-FeatureFusion: A general framework for fusing heterogeneous features via particle swarm optimization.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf263
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf263
🔥 Now published in Bioinformatics Advances: “Shiny-Calorie: A context-aware application for indirect calorimetry data analysis and visualization using R.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf270
Authors include: @wachtenlab.bsky.social, @ecsignalmetabolism.bsky.social
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf270
Authors include: @wachtenlab.bsky.social, @ecsignalmetabolism.bsky.social

November 5, 2025 at 10:01 AM
🔥 Now published in Bioinformatics Advances: “Shiny-Calorie: A context-aware application for indirect calorimetry data analysis and visualization using R.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf270
Authors include: @wachtenlab.bsky.social, @ecsignalmetabolism.bsky.social
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf270
Authors include: @wachtenlab.bsky.social, @ecsignalmetabolism.bsky.social
💊 Now published in Bioinformatics Advances: “DSA-DeepFM: A dual-stage attention-enhanced DeepFM model for predicting anticancer synergistic drug combinations.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf269
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf269

November 4, 2025 at 10:02 AM
💊 Now published in Bioinformatics Advances: “DSA-DeepFM: A dual-stage attention-enhanced DeepFM model for predicting anticancer synergistic drug combinations.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf269
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf269
🧬 Just out in Bioinformatics Advances: “TidyGWAS: A scalable approach for standardized cleaning of genome-wide association study summary statistics.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf262
Authors include: @joellepasman.bsky.social
Explore the full study: https://doi.org/10.1093/bioadv/vbaf262
Authors include: @joellepasman.bsky.social

November 3, 2025 at 10:01 AM
🧬 Just out in Bioinformatics Advances: “TidyGWAS: A scalable approach for standardized cleaning of genome-wide association study summary statistics.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf262
Authors include: @joellepasman.bsky.social
Explore the full study: https://doi.org/10.1093/bioadv/vbaf262
Authors include: @joellepasman.bsky.social
🧠 Now published in Bioinformatics Advances: “Gaining insights into Alzheimer’s disease by predicting chromatin spatial organization.”
Read the full paper here: ttps://doi.org/10.1093/bioadv/vbaf268
Read the full paper here: ttps://doi.org/10.1093/bioadv/vbaf268

October 31, 2025 at 10:02 AM
🧠 Now published in Bioinformatics Advances: “Gaining insights into Alzheimer’s disease by predicting chromatin spatial organization.”
Read the full paper here: ttps://doi.org/10.1093/bioadv/vbaf268
Read the full paper here: ttps://doi.org/10.1093/bioadv/vbaf268
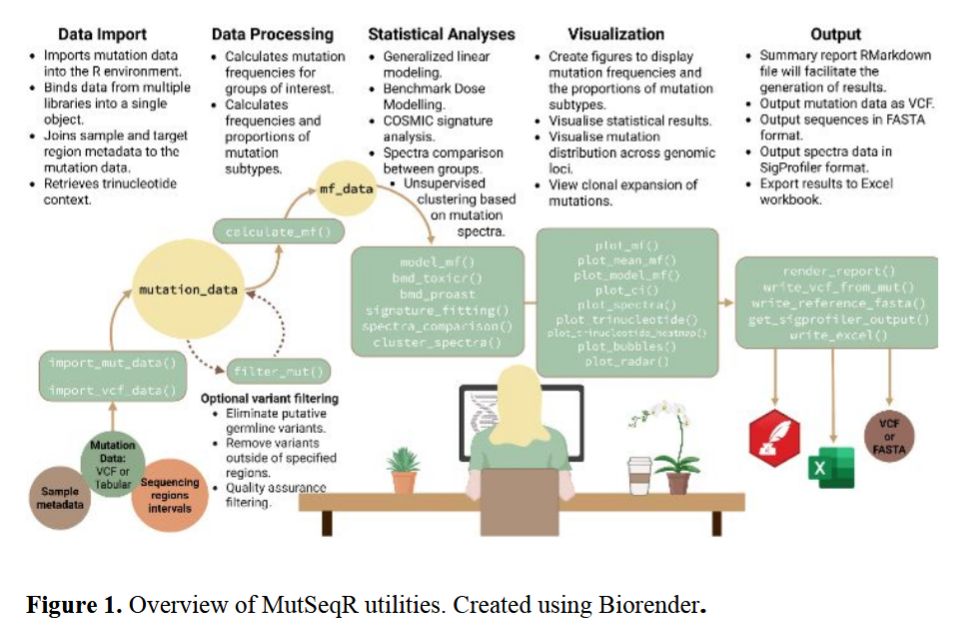
🧬 Explore the latest from Bioinformatics Advances: “MutSeqR: An open-source R package for standardized analysis of error-corrected next-generation sequencing data in genetic toxicology.”
Full article available: https://doi.org/10.1093/bioadv/vbaf265
Full article available: https://doi.org/10.1093/bioadv/vbaf265
October 31, 2025 at 9:02 AM
🧬 Explore the latest from Bioinformatics Advances: “MutSeqR: An open-source R package for standardized analysis of error-corrected next-generation sequencing data in genetic toxicology.”
Full article available: https://doi.org/10.1093/bioadv/vbaf265
Full article available: https://doi.org/10.1093/bioadv/vbaf265
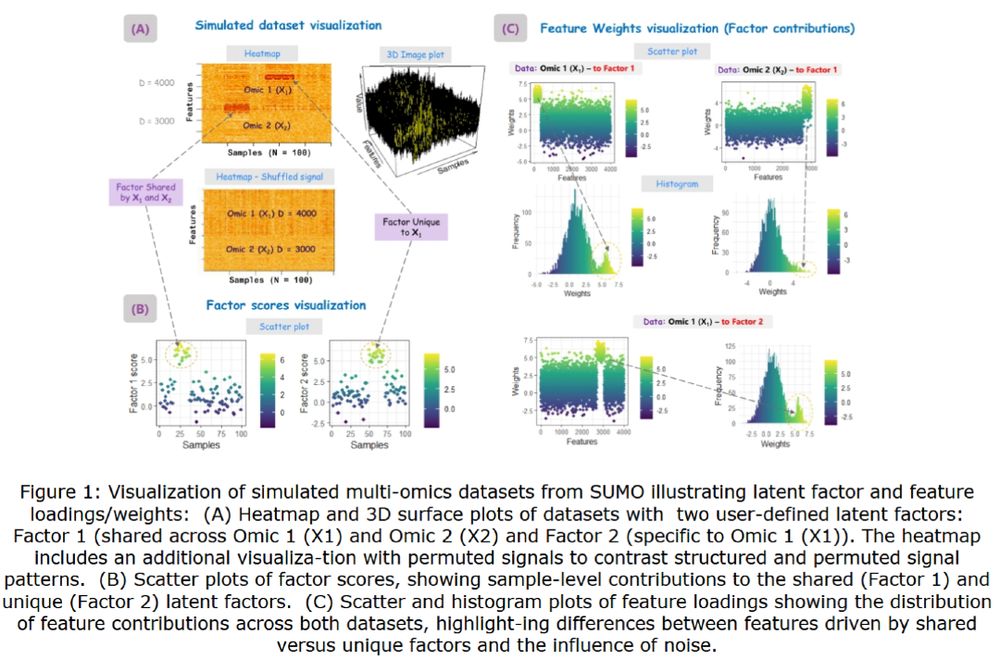
🧪 Now published in Bioinformatics Advances: “SUMO: An R package for simulating multi-omics data for methods development and testing.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf264
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf264
October 30, 2025 at 10:02 AM
🧪 Now published in Bioinformatics Advances: “SUMO: An R package for simulating multi-omics data for methods development and testing.”
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf264
Read the full paper here: https://doi.org/10.1093/bioadv/vbaf264
🧬 Just out in Bioinformatics Advances: “Unifying proteomic technologies with ProteinProjector.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf266
Explore the full study: https://doi.org/10.1093/bioadv/vbaf266

October 30, 2025 at 9:02 AM
🧬 Just out in Bioinformatics Advances: “Unifying proteomic technologies with ProteinProjector.”
Explore the full study: https://doi.org/10.1093/bioadv/vbaf266
Explore the full study: https://doi.org/10.1093/bioadv/vbaf266
🧩 Explore the latest from Bioinformatics Advances: “StarPepWeb: An integrative, graph-based resource for bioactive peptides.”
Full article available: https://doi.org/10.1093/bioadv/vbaf261
Full article available: https://doi.org/10.1093/bioadv/vbaf261

October 29, 2025 at 10:01 AM
🧩 Explore the latest from Bioinformatics Advances: “StarPepWeb: An integrative, graph-based resource for bioactive peptides.”
Full article available: https://doi.org/10.1093/bioadv/vbaf261
Full article available: https://doi.org/10.1093/bioadv/vbaf261