



Yun S. Song
@yun-s-song.bsky.social
Professor of EECS and Statistics at UC Berkeley. Mathematical and computational biologist.
To show that GPN-Star is a robust and generalizable framework that can advance biology beyond human genetics, we apply it to train gLMs for five well-studied model organisms and demonstrate their effectiveness in assessing variant effects in these species.
(8/n)
(8/n)

September 22, 2025 at 5:29 AM
To show that GPN-Star is a robust and generalizable framework that can advance biology beyond human genetics, we apply it to train gLMs for five well-studied model organisms and demonstrate their effectiveness in assessing variant effects in these species.
(8/n)
(8/n)
In addition, GPN-Star exhibits meaningful nucleotide dependencies that align with known functional dependencies, indicating its potential to help understand genomic syntax. This represents a notable advance over traditional conservation scores.
(7/n)
(7/n)

September 22, 2025 at 5:29 AM
In addition, GPN-Star exhibits meaningful nucleotide dependencies that align with known functional dependencies, indicating its potential to help understand genomic syntax. This represents a notable advance over traditional conservation scores.
(7/n)
(7/n)
By training GPN-Star on vertebrate, mammal, and primate alignments, we reveal task-dependent advantages of modeling deeper versus more recent evolution. These findings offer new biological insights and practical guidance for developing future gLMs and evolutionary models.
(6/n)
(6/n)


September 22, 2025 at 5:29 AM
By training GPN-Star on vertebrate, mammal, and primate alignments, we reveal task-dependent advantages of modeling deeper versus more recent evolution. These findings offer new biological insights and practical guidance for developing future gLMs and evolutionary models.
(6/n)
(6/n)
GPN-Star achieves unprecedented SNP heritability enrichments across over 100 human complex traits. Moreover, we devise a simple approach to incorporate tissue-specificity into the model prediction and show that it further improves heritability enrichment.
(5/n)
(5/n)

September 22, 2025 at 5:29 AM
GPN-Star achieves unprecedented SNP heritability enrichments across over 100 human complex traits. Moreover, we devise a simple approach to incorporate tissue-specificity into the model prediction and show that it further improves heritability enrichment.
(5/n)
(5/n)
We compare GPN-Star with several models, including the recent AlphaGenome and Evo2 models with up to 1Mb context size and 40B parameters, and observe that GPN-Star consistently ranks at the top across a wide range of human variant effect prediction tasks.
(4/n)
(4/n)

September 22, 2025 at 5:29 AM
We compare GPN-Star with several models, including the recent AlphaGenome and Evo2 models with up to 1Mb context size and 40B parameters, and observe that GPN-Star consistently ranks at the top across a wide range of human variant effect prediction tasks.
(4/n)
(4/n)
GPN-Star features a novel phylogeny-aware architecture that enables the model to explicitly capture evolutionary relationships encoded in whole-genome alignments and overcomes the key limitations of our earlier model GPN-MSA (doi.org/10.1038/s415...).
(2/n)
(2/n)

September 22, 2025 at 5:29 AM
GPN-Star features a novel phylogeny-aware architecture that enables the model to explicitly capture evolutionary relationships encoded in whole-genome alignments and overcomes the key limitations of our earlier model GPN-MSA (doi.org/10.1038/s415...).
(2/n)
(2/n)
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)

September 22, 2025 at 5:29 AM
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)
We interpret what sequence features the model associates with dysfunction. One example is shown below. For a specific light chain V- and J- gene, we observe sharp selection on CDRL3 length, and on certain amino acids. (9/n)

August 15, 2025 at 1:17 PM
We interpret what sequence features the model associates with dysfunction. One example is shown below. For a specific light chain V- and J- gene, we observe sharp selection on CDRL3 length, and on certain amino acids. (9/n)
In new data, we find that very low scores are associated with reduced surface expression in naive B cells. To our knowledge, this is the first time expression variation in naive B cells has been linked to the light chain. (8/n)

August 15, 2025 at 1:17 PM
In new data, we find that very low scores are associated with reduced surface expression in naive B cells. To our knowledge, this is the first time expression variation in naive B cells has been linked to the light chain. (8/n)
B cells can further mutate antibodies to improve binding. We compare observed mutations to random control sets of mutations. Mutations that significantly decrease model scores appear to be selected out. However, this only works in a few positions. (7/n)


August 15, 2025 at 1:17 PM
B cells can further mutate antibodies to improve binding. We compare observed mutations to random control sets of mutations. Mutations that significantly decrease model scores appear to be selected out. However, this only works in a few positions. (7/n)
Models trained on allelic inclusion generalize to predict antibody properties with no direct training. Here we apply models to independent data measuring polyreactivity of human antibodies and observe correlation with polyreactivity. Baselines don’t capture this signal. (6/n)


August 15, 2025 at 1:17 PM
Models trained on allelic inclusion generalize to predict antibody properties with no direct training. Here we apply models to independent data measuring polyreactivity of human antibodies and observe correlation with polyreactivity. Baselines don’t capture this signal. (6/n)
We don’t know which sequence in each double-light B cell is “bad”, but we develop a training framework that doesn’t need this information. We compare with baseline approaches that don’t use the new allelic inclusion data. (5/n)

August 15, 2025 at 1:17 PM
We don’t know which sequence in each double-light B cell is “bad”, but we develop a training framework that doesn’t need this information. We compare with baseline approaches that don’t use the new allelic inclusion data. (5/n)
We propose using double-light B cells as negative examples for antibody machine learning. Double-light B cells can be observed at scale in some recent datasets of human antibodies. Each such cell has one “bad” sequence, whereas other cells all have functional antibodies. (4/n)

August 15, 2025 at 1:17 PM
We propose using double-light B cells as negative examples for antibody machine learning. Double-light B cells can be observed at scale in some recent datasets of human antibodies. Each such cell has one “bad” sequence, whereas other cells all have functional antibodies. (4/n)
Most mature B cells express only the final, successful heavy and light chains (allelic exclusion). However, ~1% express two light chains (allelic inclusion). Previous work in mice has found that when this occurs, one of the light chains is dysfunctional. (3/n)

August 15, 2025 at 1:17 PM
Most mature B cells express only the final, successful heavy and light chains (allelic exclusion). However, ~1% express two light chains (allelic inclusion). Previous work in mice has found that when this occurs, one of the light chains is dysfunctional. (3/n)
Natural antibodies are generated in B cells and tested for function (sufficient expression, low autoreactivity). If either the heavy or light chain fails, the B cell can try to generate it again. We usually can only sequence B cells that have passed all checkpoints. (2/n)

August 15, 2025 at 1:17 PM
Natural antibodies are generated in B cells and tested for function (sufficient expression, low autoreactivity). If either the heavy or light chain fails, the B cell can try to generate it again. We usually can only sequence B cells that have passed all checkpoints. (2/n)
Specifically, we prove that, for any multi-type BDMS model, there exists a mathematically equivalent model with complete sampling and no death; we call this the Forward-Equivalent (FE) model. Our proof is constructive and hence we can find an explicit equivalent model.
6/n
6/n
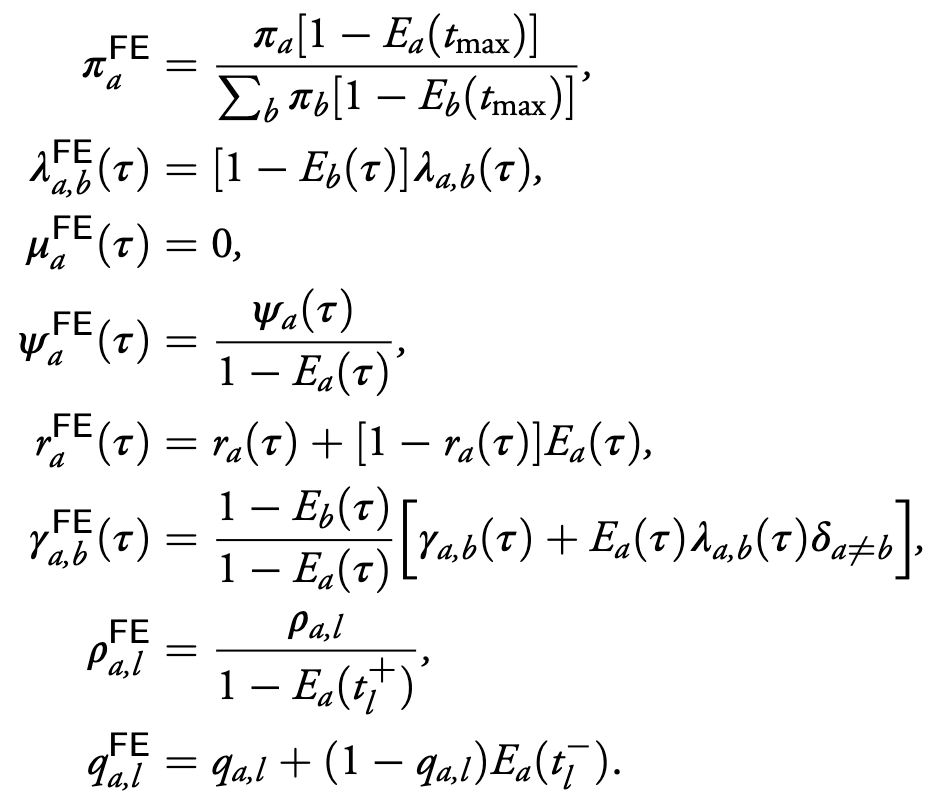
May 23, 2025 at 9:02 PM
Specifically, we prove that, for any multi-type BDMS model, there exists a mathematically equivalent model with complete sampling and no death; we call this the Forward-Equivalent (FE) model. Our proof is constructive and hence we can find an explicit equivalent model.
6/n
6/n
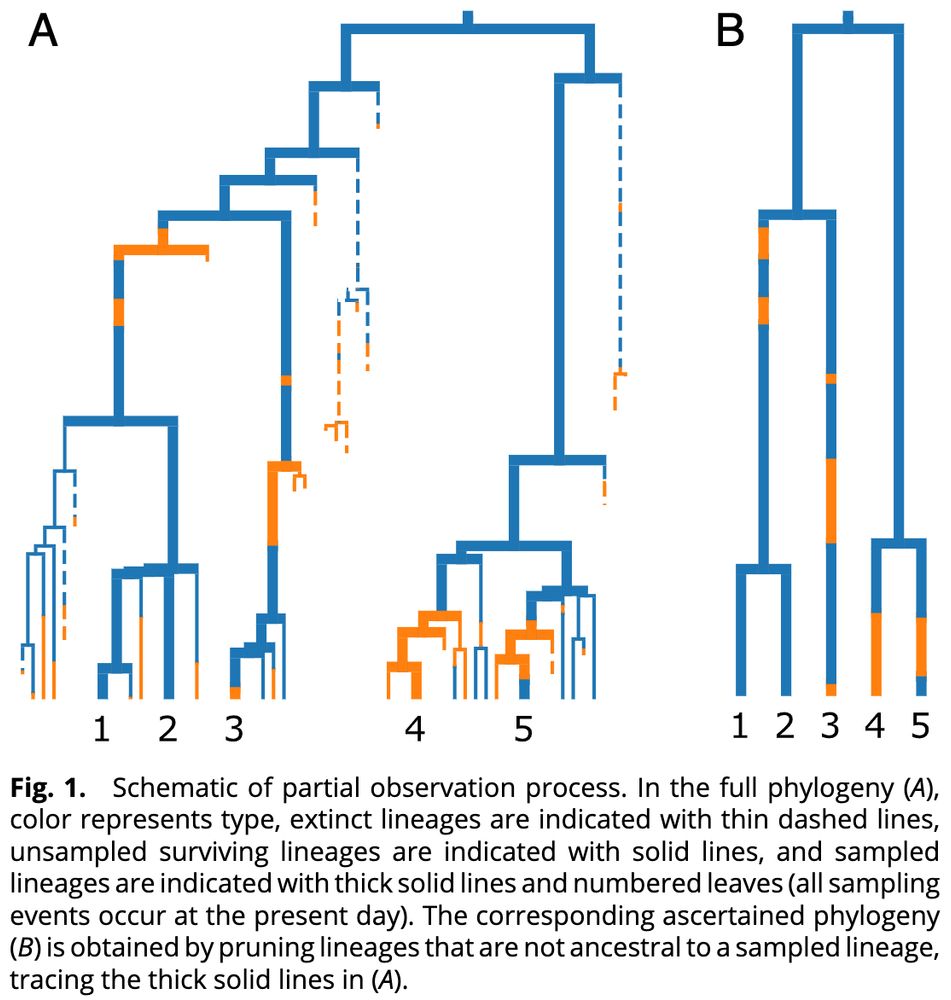
Developing and benchmarking inference methods relies on extensive tree simulation, but due to death and incomplete sampling in BDMS models, the tree describing the ancestral relationship of an observed sample represents only a partial history of the full population.
3/n
3/n
May 23, 2025 at 9:02 PM
Developing and benchmarking inference methods relies on extensive tree simulation, but due to death and incomplete sampling in BDMS models, the tree describing the ancestral relationship of an observed sample represents only a partial history of the full population.
3/n
3/n
How can one efficiently simulate phylodynamics for populations with billions of individuals, as is typical in many applications, e.g., viral evolution and cancer genomics? In this work with M. Celentano, @wsdewitt.github.io , & S. Prillo, we provide a solution. doi.org/10.1073/pnas...
1/n
1/n

May 23, 2025 at 9:02 PM
How can one efficiently simulate phylodynamics for populations with billions of individuals, as is typical in many applications, e.g., viral evolution and cancer genomics? In this work with M. Celentano, @wsdewitt.github.io , & S. Prillo, we provide a solution. doi.org/10.1073/pnas...
1/n
1/n
Thrilled to see my digital art on the cover of Trends Genet. The two binary strings represent reverse-complementary DNA sequences (00=A, 01=C, 10=G, 11=T) and the connecting rectangles represent “embeddings” learned by DNA language models. Pls check out our article as well: doi.org/10.1016/j.ti...

April 7, 2025 at 3:01 PM
Thrilled to see my digital art on the cover of Trends Genet. The two binary strings represent reverse-complementary DNA sequences (00=A, 01=C, 10=G, 11=T) and the connecting rectangles represent “embeddings” learned by DNA language models. Pls check out our article as well: doi.org/10.1016/j.ti...
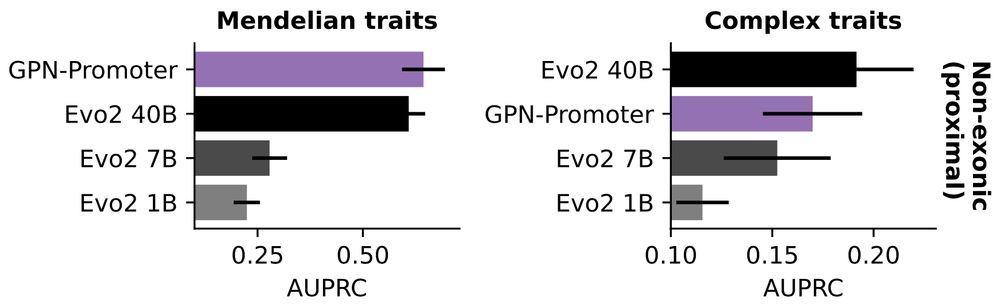
We also find that GPN-Promoter (152M params) performs comparably to Evo 2 40B when evaluating only on promoters, suggesting that if you only care about one region, you need vastly fewer parameters.
4/n
4/n
March 4, 2025 at 7:54 PM
We also find that GPN-Promoter (152M params) performs comparably to Evo 2 40B when evaluating only on promoters, suggesting that if you only care about one region, you need vastly fewer parameters.
4/n
4/n
We inspected the Evo 2 logo at the famous ZRS enhancer with evidence of functionality from conservation, ENCODE biochem assays, and disease associations (polydactyly). While Evo 2 picks up a nearby exon, it does not predict constraint on ZRS.
Browser: genome.ucsc.edu/s/gbenegas/e...
3/n
Browser: genome.ucsc.edu/s/gbenegas/e...
3/n

March 4, 2025 at 7:54 PM
We inspected the Evo 2 logo at the famous ZRS enhancer with evidence of functionality from conservation, ENCODE biochem assays, and disease associations (polydactyly). While Evo 2 picks up a nearby exon, it does not predict constraint on ZRS.
Browser: genome.ucsc.edu/s/gbenegas/e...
3/n
Browser: genome.ucsc.edu/s/gbenegas/e...
3/n
It seems that scaling substantially improves the performance of Evo 2 for promoters and ncRNA but not for distal enhancer-like elements, which were not part of its data curation process. 2/n

March 4, 2025 at 7:54 PM
It seems that scaling substantially improves the performance of Evo 2 for promoters and ncRNA but not for distal enhancer-like elements, which were not part of its data curation process. 2/n
In our updated TraitGym preprint (w/ @gonzalobenegas.bsky.social & Gökcen Eraslan), we evaluate Evo 2 on regulatory variants associated with human traits. We see marked performance gains with scale on Mendelian traits, although still a bit behind alignment-based methods.
doi.org/10.1101/2025...
1/n
doi.org/10.1101/2025...
1/n
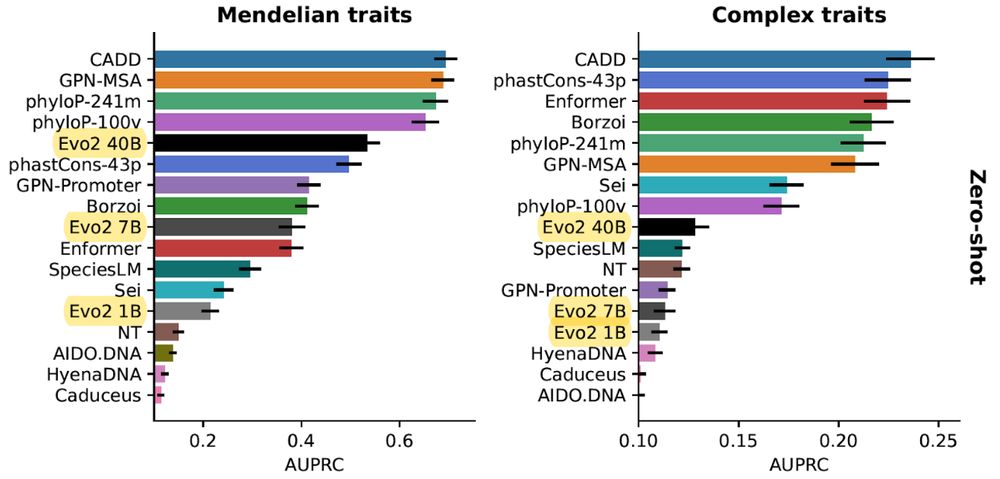
March 4, 2025 at 7:54 PM
In our updated TraitGym preprint (w/ @gonzalobenegas.bsky.social & Gökcen Eraslan), we evaluate Evo 2 on regulatory variants associated with human traits. We see marked performance gains with scale on Mendelian traits, although still a bit behind alignment-based methods.
doi.org/10.1101/2025...
1/n
doi.org/10.1101/2025...
1/n
Strikingly, on variant effect prediction, SiteRM outperforms pLMs and other models that incorporate epistatic interactions. We attribute SiteRM’s increased performance to conceptual advances in our probabilistic treatment of evolutionary data and our ability to handle extremely large MSAs.
5/7
5/7

November 16, 2024 at 8:42 PM
Strikingly, on variant effect prediction, SiteRM outperforms pLMs and other models that incorporate epistatic interactions. We attribute SiteRM’s increased performance to conceptual advances in our probabilistic treatment of evolutionary data and our ability to handle extremely large MSAs.
5/7
5/7
By leveraging the unprecedented scalability of our method, we develop a new, rich phylogenetic model called SiteRM, which can estimate a general site-specific rate matrix for each column of a given MSA. Our method can be applied to MSAs with hundreds of thousands to millions of sequences.
4/7
4/7

November 16, 2024 at 8:42 PM
By leveraging the unprecedented scalability of our method, we develop a new, rich phylogenetic model called SiteRM, which can estimate a general site-specific rate matrix for each column of a given MSA. Our method can be applied to MSAs with hundreds of thousands to millions of sequences.
4/7
4/7