



Mat Beale
@typemat12.bsky.social
Microbial (meta)genomics researcher @sangerinstitute
interested in genomic epidemiology and evolutionary dynamics of STIs/NTDs/AMR in complex samples. Views my own.
https://www.sanger.ac.uk/person/beale-mathew/
interested in genomic epidemiology and evolutionary dynamics of STIs/NTDs/AMR in complex samples. Views my own.
https://www.sanger.ac.uk/person/beale-mathew/
This is a fun diversion, but not sure it makes much sense...

November 20, 2025 at 9:31 AM
This is a fun diversion, but not sure it makes much sense...
Enjoying the beautiful weather in beautiful Montreal ahead of #stihiv2025

July 26, 2025 at 7:09 PM
Enjoying the beautiful weather in beautiful Montreal ahead of #stihiv2025
A tour de force in investigating and building capacity for a neglected tropical disease through genomics by @chewapreecha.bsky.social

May 23, 2025 at 10:20 AM
A tour de force in investigating and building capacity for a neglected tropical disease through genomics by @chewapreecha.bsky.social
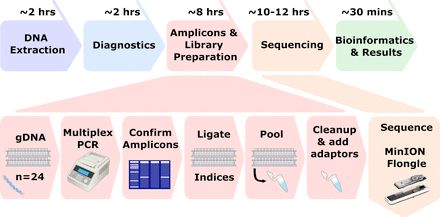
And we then deployed the method to a low-resource laboratory with our collaborators @thruzim.bsky.social BRTI in Zimbabwe as part of an ongoing genital ulcer study. The final assay can be performed in <2 days and costs <£13/sample.
February 27, 2025 at 10:12 AM
And we then deployed the method to a low-resource laboratory with our collaborators @thruzim.bsky.social BRTI in Zimbabwe as part of an ongoing genital ulcer study. The final assay can be performed in <2 days and costs <£13/sample.
We designed a 59-amplicon multiplex primer scheme which performs with high sensitivity (<qPCR Ct 32), with sequencing performed on MinION Flongle cells (24 samples per run), and high concordance between ONT amplicon SNP calls and Illumina WGS.

February 27, 2025 at 10:12 AM
We designed a 59-amplicon multiplex primer scheme which performs with high sensitivity (<qPCR Ct 32), with sequencing performed on MinION Flongle cells (24 samples per run), and high concordance between ONT amplicon SNP calls and Illumina WGS.
With novel (previously undescribed diversity), it functions more like a highly sensitive MLST - randomly occurring SNPs will occur by chance in amplicons, but we have many more amplicons than a typical MLST scheme so we have more opportunity to detect novelty.

February 27, 2025 at 10:12 AM
With novel (previously undescribed diversity), it functions more like a highly sensitive MLST - randomly occurring SNPs will occur by chance in amplicons, but we have many more amplicons than a typical MLST scheme so we have more opportunity to detect novelty.
This enables high resolution reconstruction of a WGS phylogeny using a small % of the genome (3.6% of T. pallidum genome here). The method recovers known sublineages (<20 SNPs apart) with high precision

February 27, 2025 at 10:12 AM
This enables high resolution reconstruction of a WGS phylogeny using a small % of the genome (3.6% of T. pallidum genome here). The method recovers known sublineages (<20 SNPs apart) with high precision
We can use this to identify information rich regions, and then hierarchically select the optimal combination of regions to ensure coverage of all sublineages. We demonstrate this using a 59-amplicon scheme for Treponema pallidum (causative agent of syphilis)

February 27, 2025 at 10:12 AM
We can use this to identify information rich regions, and then hierarchically select the optimal combination of regions to ensure coverage of all sublineages. We demonstrate this using a 59-amplicon scheme for Treponema pallidum (causative agent of syphilis)
When we identify SNPs discriminating individual finescale sublineages (e.g. <20 SNPs apart), the SNPs are scattered along a bacterial genome. However, if we consider discriminatory SNPs for many sublineages together, by chance we find positional clustering

February 27, 2025 at 10:12 AM
When we identify SNPs discriminating individual finescale sublineages (e.g. <20 SNPs apart), the SNPs are scattered along a bacterial genome. However, if we consider discriminatory SNPs for many sublineages together, by chance we find positional clustering
Genomic pathogen surveillance is costly & challenging but multiplex amplicons eg @joshquick.bsky.social primalscheme works for viruses. Applying to larger (bacterial) genomes means compromise. For known pop structures, we identify key ancestral SNPs, then find regions which maximise discrimination

February 27, 2025 at 10:12 AM
Genomic pathogen surveillance is costly & challenging but multiplex amplicons eg @joshquick.bsky.social primalscheme works for viruses. Applying to larger (bacterial) genomes means compromise. For known pop structures, we identify key ancestral SNPs, then find regions which maximise discrimination