



Jason Sheltzer
@jsheltzer.bsky.social
Assistant prof at Stanford. Interested in aneuploidy, mitotic kinases, cancer therapeutics, and drug development. Co-founder x2.
We injected the G568S mice with a mouse cancer cell line and then treated them with a high dose of MEL-495R (which was tolerable to the G568S mice but toxic to WT mice). This resulted in a significant anti-cancer effect, verifying that on-target toxicity limits effective dosing.

August 4, 2025 at 6:27 PM
We injected the G568S mice with a mouse cancer cell line and then treated them with a high dose of MEL-495R (which was tolerable to the G568S mice but toxic to WT mice). This resulted in a significant anti-cancer effect, verifying that on-target toxicity limits effective dosing.
This gave us confidence to move forward with the drug. We identified a non-toxic dose of MEL-495R and tested it in several xenografts. However, it showed very little anti-cancer activity. A splicing qPCR indicated that this non-toxic dose wasn’t appreciably inhibiting CDK11.


August 4, 2025 at 6:27 PM
This gave us confidence to move forward with the drug. We identified a non-toxic dose of MEL-495R and tested it in several xenografts. However, it showed very little anti-cancer activity. A splicing qPCR indicated that this non-toxic dose wasn’t appreciably inhibiting CDK11.
We bred a large cohort of CDK11-mutant (G568S) and CDK11-WT mice, treated them with an ultra-high dose of MEL-495R, and it worked beautifully. The wild-type mice became very sick while the CDK11-G568S mice were totally fine. Our drug is specific for CDK11 - in living mice!

August 4, 2025 at 6:27 PM
We bred a large cohort of CDK11-mutant (G568S) and CDK11-WT mice, treated them with an ultra-high dose of MEL-495R, and it worked beautifully. The wild-type mice became very sick while the CDK11-G568S mice were totally fine. Our drug is specific for CDK11 - in living mice!
We thought - if the mice expressing this mutation are still affected by our CDK11 inhibitor, then that tells us that it’s causing CDK11-independent toxicity. In contrast, if these mice are resistant to the drug, then any side effects of the drug in WT mice are due to CDK11.

August 4, 2025 at 6:27 PM
We thought - if the mice expressing this mutation are still affected by our CDK11 inhibitor, then that tells us that it’s causing CDK11-independent toxicity. In contrast, if these mice are resistant to the drug, then any side effects of the drug in WT mice are due to CDK11.
We came up with a way to answer this question. We had discovered a mutation in CDK11 that blocks drug binding to it. We thought - what if we put that mutation into a mouse? So, we found the mouse ortholog of the human mutation, CRISPR’d it into some zygotes, and did exactly that.


August 4, 2025 at 6:27 PM
We came up with a way to answer this question. We had discovered a mutation in CDK11 that blocks drug binding to it. We thought - what if we put that mutation into a mouse? So, we found the mouse ortholog of the human mutation, CRISPR’d it into some zygotes, and did exactly that.
Now, we wanted to take the drug in vivo. Unfortunately, it had a terrible ADME profile. We worked with the talented chemists at Meliora to develop an improved CDK11 inhibitor, and we created MEL-495R, which exhibits potent CDK11 inhibition and superior PK properties.




August 4, 2025 at 6:27 PM
Now, we wanted to take the drug in vivo. Unfortunately, it had a terrible ADME profile. We worked with the talented chemists at Meliora to develop an improved CDK11 inhibitor, and we created MEL-495R, which exhibits potent CDK11 inhibition and superior PK properties.
Next, we figured out why - 1p36 is where CDK11 and its activating cyclin (cyclin L) are encoded. Having a lower dosage of these genes enhances the dependency on the remaining enzyme, creating a synthetic-lethal relationship.


August 4, 2025 at 6:27 PM
Next, we figured out why - 1p36 is where CDK11 and its activating cyclin (cyclin L) are encoded. Having a lower dosage of these genes enhances the dependency on the remaining enzyme, creating a synthetic-lethal relationship.
For any cancer therapy, finding a biomarker to predict sensitivity is key. We analyzed screening data with CDK11-targeting CRISPR, CDK11-targeting RNAi, and OTS964 treatment, and they all pointed to the same biomarker: Chr1p36 deletions enhance sensitivity to CDK11 ablation.


August 4, 2025 at 6:27 PM
For any cancer therapy, finding a biomarker to predict sensitivity is key. We analyzed screening data with CDK11-targeting CRISPR, CDK11-targeting RNAi, and OTS964 treatment, and they all pointed to the same biomarker: Chr1p36 deletions enhance sensitivity to CDK11 ablation.
Next, we uncovered the cell biology of CDK11 inhibition. We found that CDK11 controls gene expression through two distinct mechanisms: ensuring accurate splicing via SF3B1, and, separately, controlling the transcription of certain genes by promoting the activation of RNAPII.


August 4, 2025 at 6:27 PM
Next, we uncovered the cell biology of CDK11 inhibition. We found that CDK11 controls gene expression through two distinct mechanisms: ensuring accurate splicing via SF3B1, and, separately, controlling the transcription of certain genes by promoting the activation of RNAPII.
First, we did the fundamental molecular genetics: targeting CDK11 with OTS964 or CRISPR killed cancer cells, and this could be rescued with a mutation in CDK11 that blocked drug binding or with gRNA-resistant cDNA (but not if the cDNA contained a kinase-inactivating substitution).


August 4, 2025 at 6:27 PM
First, we did the fundamental molecular genetics: targeting CDK11 with OTS964 or CRISPR killed cancer cells, and this could be rescued with a mutation in CDK11 that blocked drug binding or with gRNA-resistant cDNA (but not if the cDNA contained a kinase-inactivating substitution).
One of these mischaracterized drugs is called OTS964. While it was initially developed as a PBK inhibitor, we showed that it actually functions by inhibiting CDK11 - making it the first-ever inhibitor of this poorly-characterized kinase.

August 4, 2025 at 6:27 PM
One of these mischaracterized drugs is called OTS964. While it was initially developed as a PBK inhibitor, we showed that it actually functions by inhibiting CDK11 - making it the first-ever inhibitor of this poorly-characterized kinase.
Thrilled to share our new paper describing the development and characterization of CDK11 inhibitors for cancer therapy. We also establish a new system that I think represents a huge leap forward in our ability to understand drug toxicity in a living organism.

August 4, 2025 at 6:27 PM
Thrilled to share our new paper describing the development and characterization of CDK11 inhibitors for cancer therapy. We also establish a new system that I think represents a huge leap forward in our ability to understand drug toxicity in a living organism.
At Stanford, I’ll be affiliated with the Stanford Cancer Institute, Radiation Oncology, and Pathology. My lab will be in the Research Park complex on Page Mill Road. I’m looking forward to getting to know my new community! Feel free to reach out and say hi.

June 4, 2025 at 5:52 PM
At Stanford, I’ll be affiliated with the Stanford Cancer Institute, Radiation Oncology, and Pathology. My lab will be in the Research Park complex on Page Mill Road. I’m looking forward to getting to know my new community! Feel free to reach out and say hi.
I’m thrilled to share that I’m joining the faculty at the Stanford University School of Medicine. My lab and I will be relocating to Stanford this summer!

June 4, 2025 at 5:52 PM
I’m thrilled to share that I’m joining the faculty at the Stanford University School of Medicine. My lab and I will be relocating to Stanford this summer!
For the first time since January, notification of a new NIH study section meeting has been posted on the Federal Register. It seems to cover multiple review panels, including one for the DP2/New Innovator grant.
www.federalregister.gov/agencies/nat...
www.federalregister.gov/agencies/nat...

March 4, 2025 at 2:55 PM
For the first time since January, notification of a new NIH study section meeting has been posted on the Federal Register. It seems to cover multiple review panels, including one for the DP2/New Innovator grant.
www.federalregister.gov/agencies/nat...
www.federalregister.gov/agencies/nat...
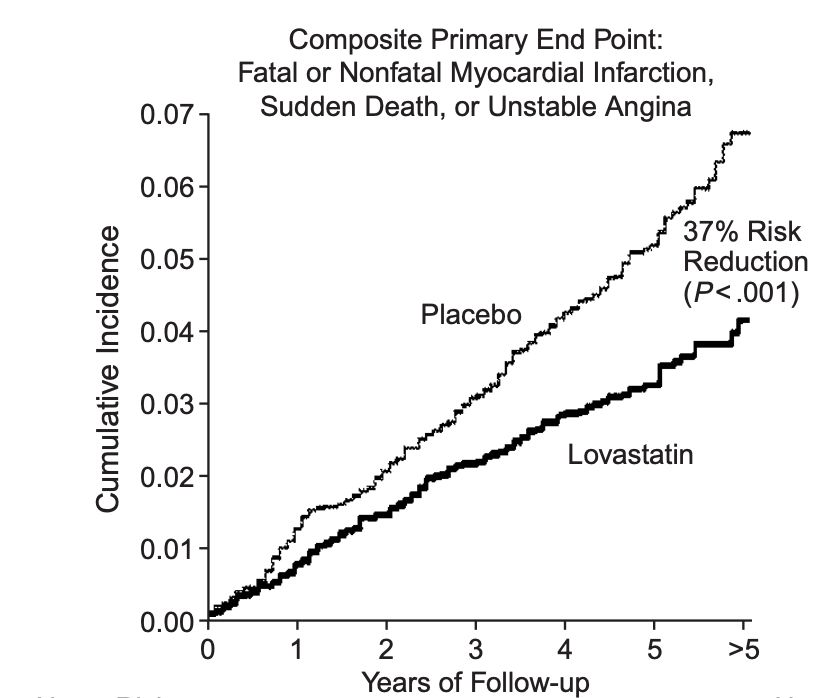
Bolstered by these findings, Merck revived its statin program and launched large-scale trials that proved that statins safely lowered the risk of heart attacks. In 1987, lovastatin earned FDA approval, launching a new era in heart disease prevention.
pmc.ncbi.nlm.nih.gov/articles/PMC...
pmc.ncbi.nlm.nih.gov/articles/PMC...
February 27, 2025 at 3:25 PM
Bolstered by these findings, Merck revived its statin program and launched large-scale trials that proved that statins safely lowered the risk of heart attacks. In 1987, lovastatin earned FDA approval, launching a new era in heart disease prevention.
pmc.ncbi.nlm.nih.gov/articles/PMC...
pmc.ncbi.nlm.nih.gov/articles/PMC...
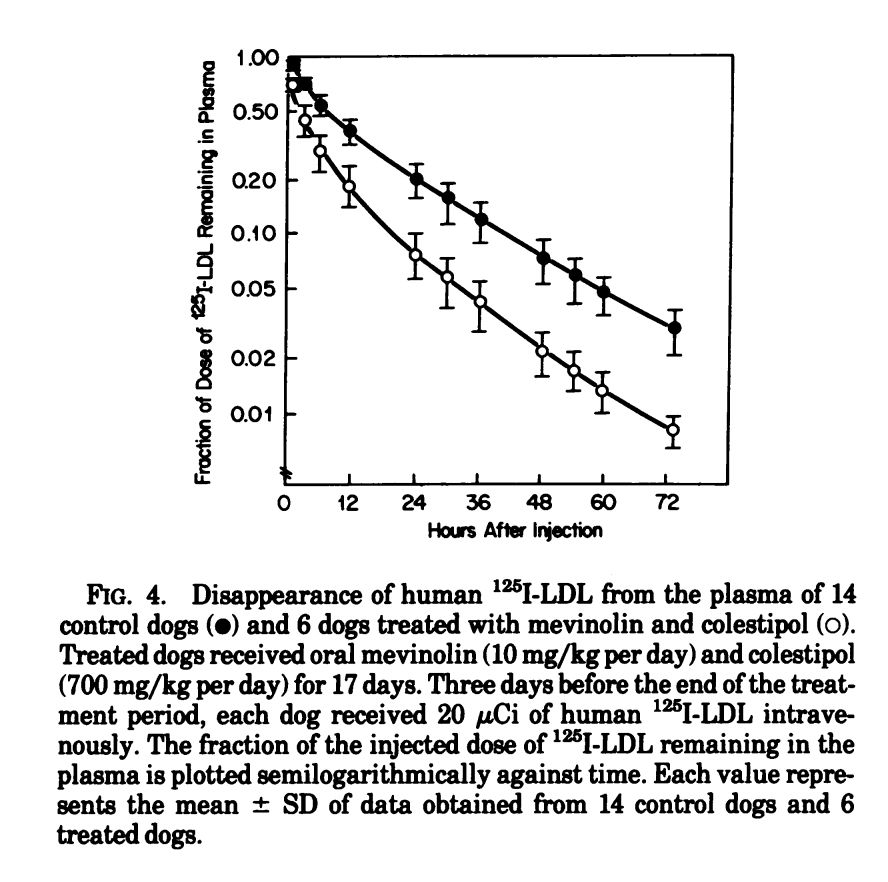
Outside of pharma, NIH-funded researchers continued to evaluate statins. Brown and Goldstein showed that it safely lowered cholesterol in dogs, and others showed that it produced positive results in patients with abnormally high cholesterol levels.
pubmed.ncbi.nlm.nih.gov/6262757/
pubmed.ncbi.nlm.nih.gov/6262757/
February 27, 2025 at 3:25 PM
Outside of pharma, NIH-funded researchers continued to evaluate statins. Brown and Goldstein showed that it safely lowered cholesterol in dogs, and others showed that it produced positive results in patients with abnormally high cholesterol levels.
pubmed.ncbi.nlm.nih.gov/6262757/
pubmed.ncbi.nlm.nih.gov/6262757/
NIH-funded investigators, including Konrad Bloch, Michael Brown, and Joseph Goldstein, uncovered the biochemical pathways that produced cholesterol and identified HMG-CoA reductase as a potential target for decreasing cholesterol synthesis.
www.sciencedirect.com/science/arti...
www.sciencedirect.com/science/arti...
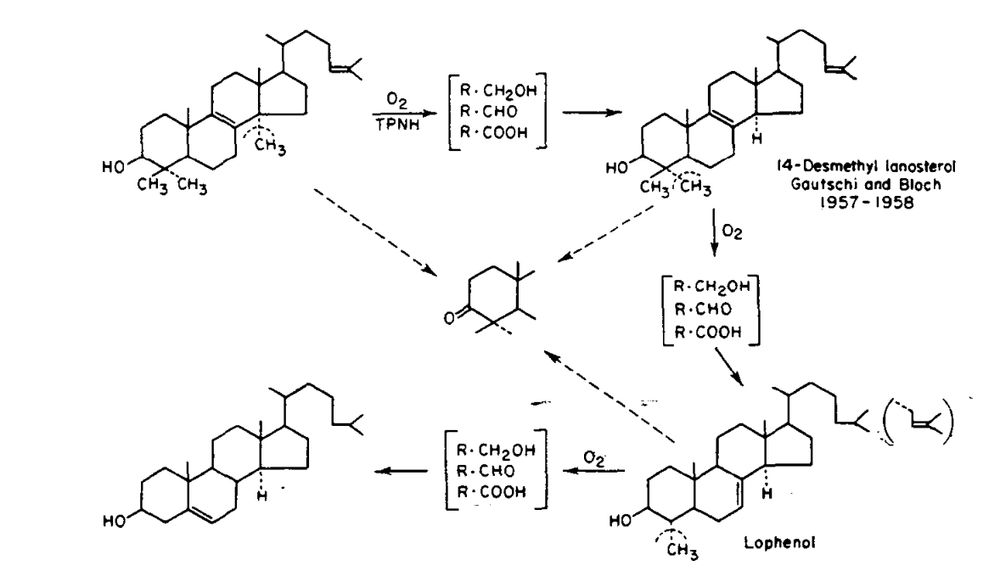
February 27, 2025 at 3:25 PM
NIH-funded investigators, including Konrad Bloch, Michael Brown, and Joseph Goldstein, uncovered the biochemical pathways that produced cholesterol and identified HMG-CoA reductase as a potential target for decreasing cholesterol synthesis.
www.sciencedirect.com/science/arti...
www.sciencedirect.com/science/arti...