



James Hemker
@jahemker.bsky.social
Stanford Dev Bio PhD
@petrovadmitri.bsky.social's lab
Carcharodontosaurus is my favorite dinosaur.
@petrovadmitri.bsky.social's lab
Carcharodontosaurus is my favorite dinosaur.
Unfortunately we haven’t had the chance to look at other organisms. I think repeat % could matter more if it starts creating large blocks of repetitive sequence (which would then need longer reads). More # of repeat elements should be ok, provided they are surrounded by unique flanking sequence.
June 21, 2025 at 9:05 PM
Unfortunately we haven’t had the chance to look at other organisms. I think repeat % could matter more if it starts creating large blocks of repetitive sequence (which would then need longer reads). More # of repeat elements should be ok, provided they are surrounded by unique flanking sequence.
Many thanks to my co-authors, @hgellert.bsky.social, Jess Smiley-Rhodes, @bernardkim.bsky.social, and @petrovadmitri.bsky.social, without whom this work would not have been possible!
April 25, 2025 at 8:03 PM
Many thanks to my co-authors, @hgellert.bsky.social, Jess Smiley-Rhodes, @bernardkim.bsky.social, and @petrovadmitri.bsky.social, without whom this work would not have been possible!
Our results suggest that reads at least 3x longer than the largest repetitive elements are required to avoid SV-calling errors from these elements. They also highlight that SV-calling is a species-specific problem, as the repeat landscape varies greatly across taxa.
April 25, 2025 at 8:03 PM
Our results suggest that reads at least 3x longer than the largest repetitive elements are required to avoid SV-calling errors from these elements. They also highlight that SV-calling is a species-specific problem, as the repeat landscape varies greatly across taxa.
Finally, we short-read sequenced our inbred lines as the vast majority of genomic data is from NGS. Unsurprisingly, short-read data had the poorest accuracy, as well as the most significant biases against insertions and the largest number of spurious inversion calls.

April 25, 2025 at 8:03 PM
Finally, we short-read sequenced our inbred lines as the vast majority of genomic data is from NGS. Unsurprisingly, short-read data had the poorest accuracy, as well as the most significant biases against insertions and the largest number of spurious inversion calls.
We additionally downsampled our 30x-coverage ultra-long reads to 20x- and 10x- coverage. We found that accuracy decreased even at 20x-coverage, and neither low-coverage distribution could recover all three of the cosmopolitan inversions.

April 25, 2025 at 8:03 PM
We additionally downsampled our 30x-coverage ultra-long reads to 20x- and 10x- coverage. We found that accuracy decreased even at 20x-coverage, and neither low-coverage distribution could recover all three of the cosmopolitan inversions.
A second significant source of error came from incorrectly merging strain-level SVs into the population call sets. While joint genotyping is well-understood for SNPs, merging SVs is still a highly complex problem that needs to rely on multiple lines of evidence to find that two SVs are the same.
April 25, 2025 at 8:03 PM
A second significant source of error came from incorrectly merging strain-level SVs into the population call sets. While joint genotyping is well-understood for SNPs, merging SVs is still a highly complex problem that needs to rely on multiple lines of evidence to find that two SVs are the same.
We were able to determine the cause of false positive SV calls from manual validation. More than half of all errors came from misalignments due to transposable elements (TEs) or complex genomic loci — except for the ultra-long distribution, which had no major issues with TEs or complex regions.
April 25, 2025 at 8:03 PM
We were able to determine the cause of false positive SV calls from manual validation. More than half of all errors came from misalignments due to transposable elements (TEs) or complex genomic loci — except for the ultra-long distribution, which had no major issues with TEs or complex regions.
When we focused on SVs > 10kb, we found that the ultra-long data was very clearly the most accurate. Only the ultra-long data had 100% accuracy when calling large inversions, finding three cosmopolitan inversions in our dataset.
April 25, 2025 at 8:03 PM
When we focused on SVs > 10kb, we found that the ultra-long data was very clearly the most accurate. Only the ultra-long data had 100% accuracy when calling large inversions, finding three cosmopolitan inversions in our dataset.
We report significant shifts in SV-calling accuracy at the population level when systematically varying read length within D. melanogaster. Our ultra-long (as defined by ONT: read N50 > 50kb) read distribution, called more SVs, and at a significantly higher accuracy, than any other distribution.

April 25, 2025 at 8:03 PM
We report significant shifts in SV-calling accuracy at the population level when systematically varying read length within D. melanogaster. Our ultra-long (as defined by ONT: read N50 > 50kb) read distribution, called more SVs, and at a significantly higher accuracy, than any other distribution.
As no definitive benchmark SV call sets exist for D. melanogaster, we then manually validated more 2,300 SVs at over 18,000 genomic loci across the read-length distributions to assess variant-calling accuracy. Validation was done by visualizing read alignments in Jbrowse2.

April 25, 2025 at 8:03 PM
As no definitive benchmark SV call sets exist for D. melanogaster, we then manually validated more 2,300 SVs at over 18,000 genomic loci across the read-length distributions to assess variant-calling accuracy. Validation was done by visualizing read alignments in Jbrowse2.
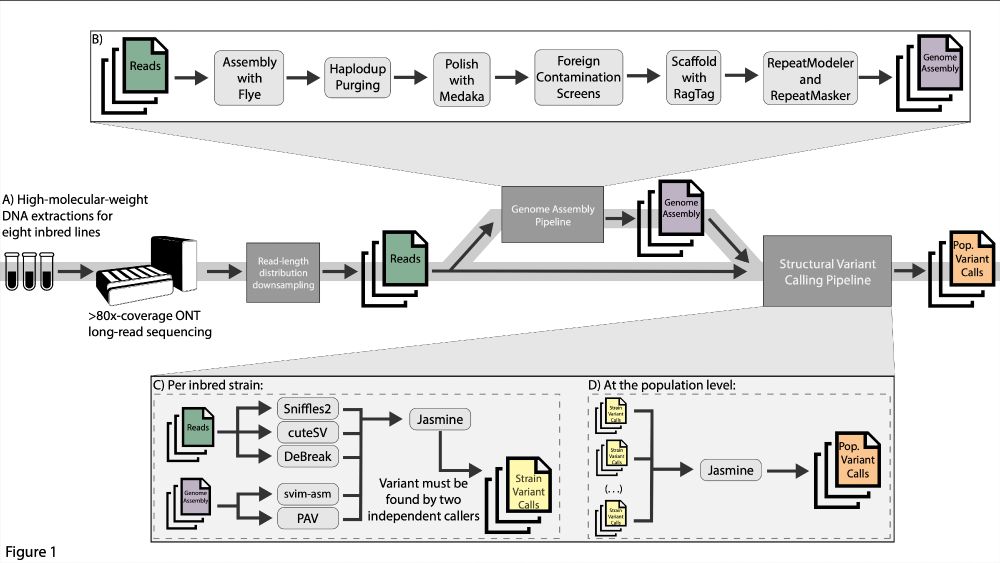
Using a combination of read-based (Sniffles2, cuteSV, DeBreak) and assembly-based callers (svim-asm, PAV), we called structural variants in every line and read-length distribution. Each strain's calls were merged together (Jasmine) for each distribution, creating a pop. level variant call set.
April 25, 2025 at 8:03 PM
Using a combination of read-based (Sniffles2, cuteSV, DeBreak) and assembly-based callers (svim-asm, PAV), we called structural variants in every line and read-length distribution. Each strain's calls were merged together (Jasmine) for each distribution, creating a pop. level variant call set.
To investigate this, we Nanopore sequenced eight D. melanogaster inbred lines to extremely high coverage (mean 238x) and then downsampled the reads to create 30x-coverage pools of distinct read-length distributions (as quantified by read N50). We additionally assembled genomes for each pool.
April 25, 2025 at 8:03 PM
To investigate this, we Nanopore sequenced eight D. melanogaster inbred lines to extremely high coverage (mean 238x) and then downsampled the reads to create 30x-coverage pools of distinct read-length distributions (as quantified by read N50). We additionally assembled genomes for each pool.
Just as a drastic impact on structural variant calling was seen moving from short reads to long reads, it is likely that the accuracy of structural variant calling significantly varies along the spectrum of read lengths produced by long-read sequencing methods.
April 25, 2025 at 8:03 PM
Just as a drastic impact on structural variant calling was seen moving from short reads to long reads, it is likely that the accuracy of structural variant calling significantly varies along the spectrum of read lengths produced by long-read sequencing methods.
@nanoporetech.com long-reads can range 1000-fold in length (100s bp - 1Mb) in a single sequencing run, but the consequences of significantly different long-read lengths on the accuracy of genome-wide structural variant calling is not well understood, especially for non-human species.
April 25, 2025 at 8:03 PM
@nanoporetech.com long-reads can range 1000-fold in length (100s bp - 1Mb) in a single sequencing run, but the consequences of significantly different long-read lengths on the accuracy of genome-wide structural variant calling is not well understood, especially for non-human species.
The increasing availability of long-read data is leading to a boom in population-level SV datasets. These datasets are critical for uncovering SV polymorphisms, which better capture the dynamic evolutionary processes that shape SV diversity.
April 25, 2025 at 8:03 PM
The increasing availability of long-read data is leading to a boom in population-level SV datasets. These datasets are critical for uncovering SV polymorphisms, which better capture the dynamic evolutionary processes that shape SV diversity.