




Ben Orr
@benorr.bsky.social
PhD Candidate, UCSF Biophysics, Kortemme Lab
Computational Biology & AI Lead, Animate Bio
ML for Protein Design
Computational Biology & AI Lead, Animate Bio
ML for Protein Design
Model weights have been uploaded to zenodo. Fine-tuning and analysis code to be released soon. Work by @benorr.bsky.social, Stephanie Crilly, @dakpinaroglu.bsky.social, Eleanor Zhu, Michael Keiser, and Tanja Kortemme. (9/9) zenodo.org/records/1558...
An improved model for prediction of de novo designed proteins with diverse geometries
Fine-tuned AF2 models for "Benjamin Orr*, Stephanie E. Crilly*, Deniz Akpinaroglu, Eleanor Zhu, Michael J. Keiser, Tanja Kortemme. An improved model for prediction of de novo designed proteins with di...
zenodo.org
June 10, 2025 at 6:28 PM
Model weights have been uploaded to zenodo. Fine-tuning and analysis code to be released soon. Work by @benorr.bsky.social, Stephanie Crilly, @dakpinaroglu.bsky.social, Eleanor Zhu, Michael Keiser, and Tanja Kortemme. (9/9) zenodo.org/records/1558...
This work highlights how augmenting existing models with informative experimental data, as presented here, could expand our exploration of designable protein space and ultimately enable more challenging design problems to be addressed than currently possible. (8/9)
June 10, 2025 at 6:28 PM
This work highlights how augmenting existing models with informative experimental data, as presented here, could expand our exploration of designable protein space and ultimately enable more challenging design problems to be addressed than currently possible. (8/9)
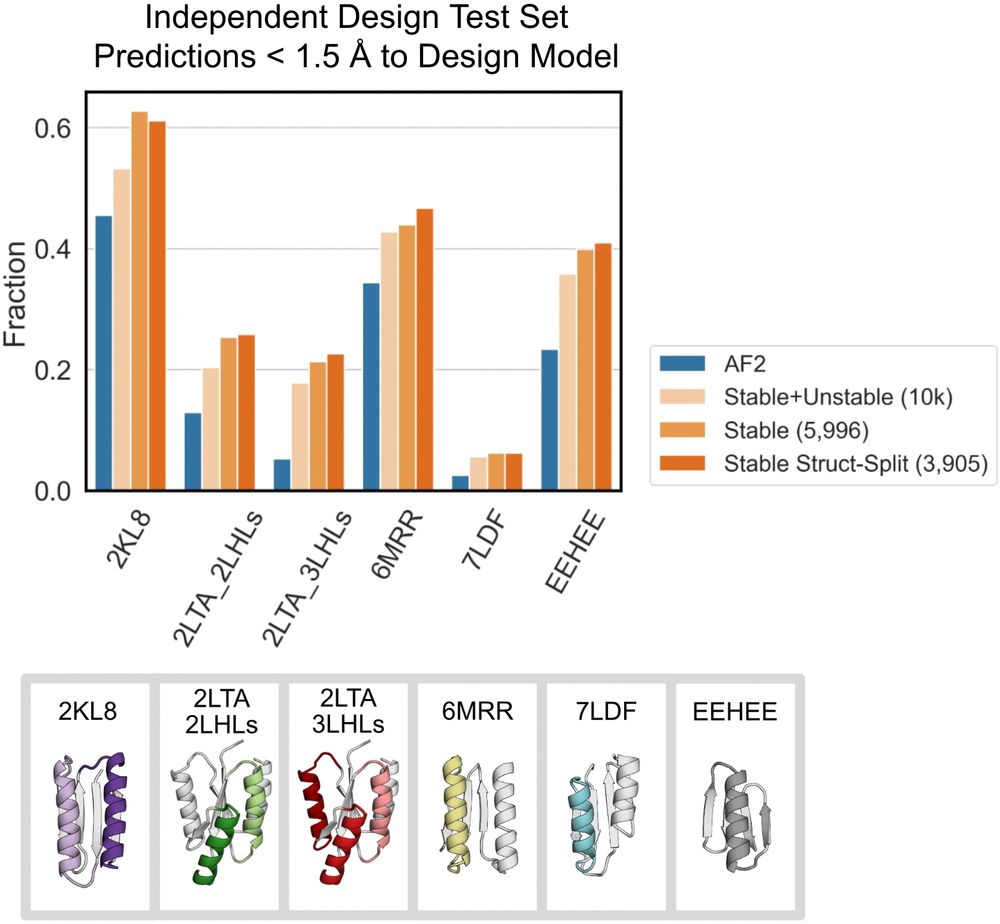
Fine-tuning AF2 on the stable sequences’ Rosetta models improves predictions for geometrically diverse proteins across 5 protein folds. Fine-tuning on ~6k stable designs leads to better performance than fine-tuning on all 10k stable+unstable designs. (7/9)
June 10, 2025 at 6:28 PM
Fine-tuning AF2 on the stable sequences’ Rosetta models improves predictions for geometrically diverse proteins across 5 protein folds. Fine-tuning on ~6k stable designs leads to better performance than fine-tuning on all 10k stable+unstable designs. (7/9)
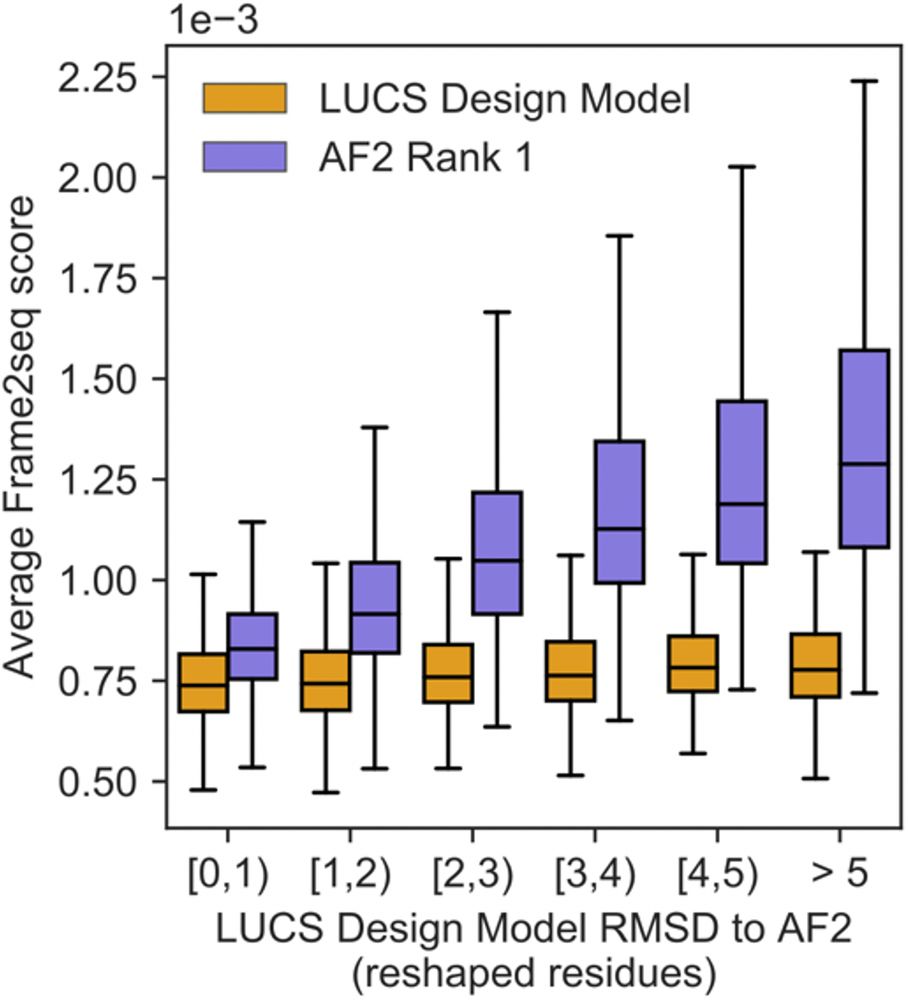
Frame2seq [@dakpinaroglu.bsky.social 2023] scores higher sequence-structure compatibility for the Rosetta models than the AF2 predictions for these stable designs, suggesting that the Rosetta models are more accurate structures than the AF2 predictions for these sequences. (6/9)
June 10, 2025 at 6:28 PM
Frame2seq [@dakpinaroglu.bsky.social 2023] scores higher sequence-structure compatibility for the Rosetta models than the AF2 predictions for these stable designs, suggesting that the Rosetta models are more accurate structures than the AF2 predictions for these sequences. (6/9)
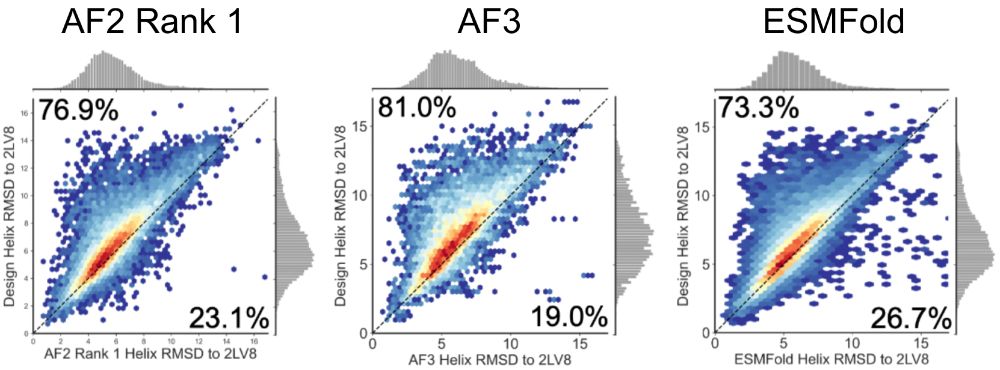
We extended this analysis to 10k diverse Rossmann fold proteins generated by LUCS and tested for stability using yeast display [@grocklin.bsky.social 2017]. For ~6k stable designs, AF2, AF3, and ESMFold all demonstrate a strong bias toward predicting more “idealized” helix geometries. (5/9)
June 10, 2025 at 6:28 PM
We extended this analysis to 10k diverse Rossmann fold proteins generated by LUCS and tested for stability using yeast display [@grocklin.bsky.social 2017]. For ~6k stable designs, AF2, AF3, and ESMFold all demonstrate a strong bias toward predicting more “idealized” helix geometries. (5/9)
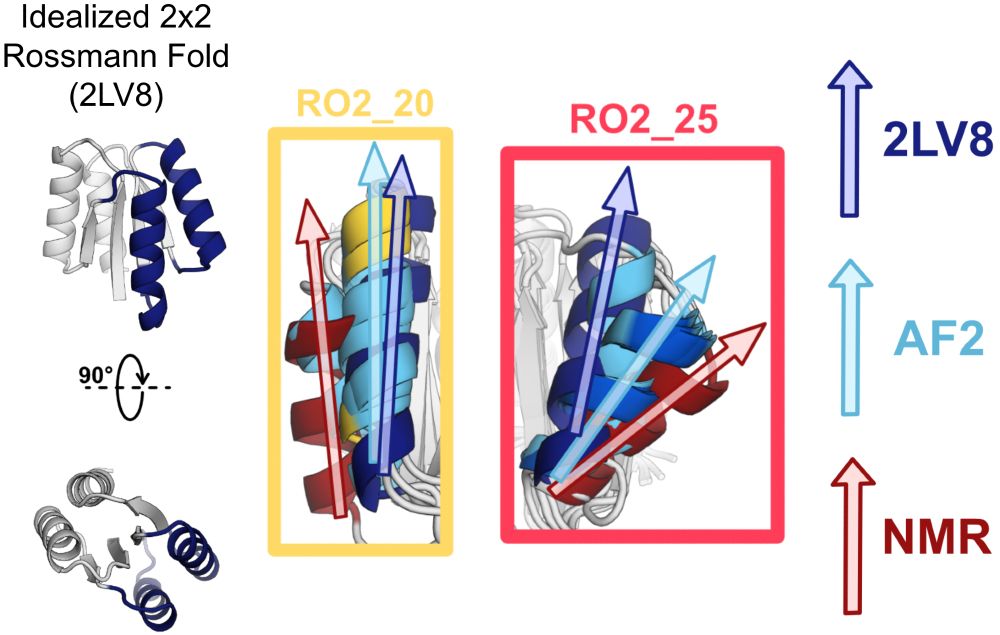
We asked whether protein structure prediction models are biased toward idealized structures for de novo proteins. Indeed, for de novo proteins with diverse geometries, AlphaFold2 predicts structures closer to an idealized de novo protein than the solved NMR structures. (4/9)
June 10, 2025 at 6:28 PM
We asked whether protein structure prediction models are biased toward idealized structures for de novo proteins. Indeed, for de novo proteins with diverse geometries, AlphaFold2 predicts structures closer to an idealized de novo protein than the solved NMR structures. (4/9)
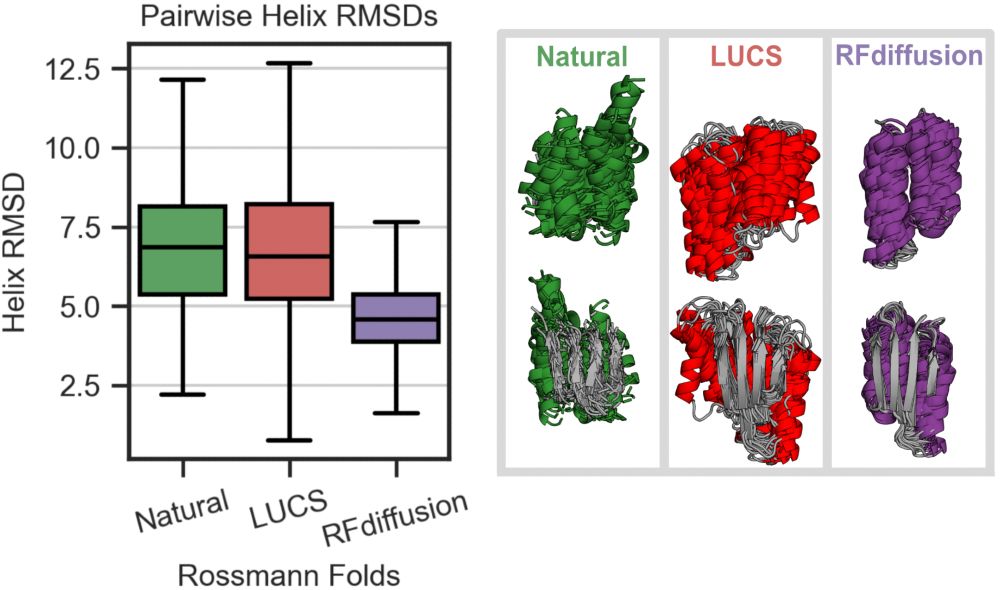
We find that a physics-based method (LUCS) samples greater structural diversity, approaching that observed in natural proteins, in a model protein fold than RFdiffusion, a generative model which utilizes the deep learning-based structure prediction network RoseTTAFold. (3/9)
June 10, 2025 at 6:28 PM
We find that a physics-based method (LUCS) samples greater structural diversity, approaching that observed in natural proteins, in a model protein fold than RFdiffusion, a generative model which utilizes the deep learning-based structure prediction network RoseTTAFold. (3/9)
In this work we explored how deep learning methods for structure prediction and design may limit our exploration of designable protein space, by favoring “idealized” structures for de novo proteins, and how to overcome these limitations with new data and improved models. (2/9)
June 10, 2025 at 6:28 PM
In this work we explored how deep learning methods for structure prediction and design may limit our exploration of designable protein space, by favoring “idealized” structures for de novo proteins, and how to overcome these limitations with new data and improved models. (2/9)