



Ryan Hisner
@ryanhisner.bsky.social
Teacher. Learner. Investigating mysteries of SARS-CoV-2 evolution. LongDesertTrain on another platform.
Already two more BA.3.2.2 from Ireland today, both closely related to the first (and both with the ORF1a:∆141-143 deletion also found in most European BA.3.2.1).
Only real change was N:T271S in one of the two. (A29086T was inherited, but C->G muts like this are very rare.)
Only real change was N:T271S in one of the two. (A29086T was inherited, but C->G muts like this are very rare.)

November 28, 2025 at 7:58 PM
Already two more BA.3.2.2 from Ireland today, both closely related to the first (and both with the ORF1a:∆141-143 deletion also found in most European BA.3.2.1).
Only real change was N:T271S in one of the two. (A29086T was inherited, but C->G muts like this are very rare.)
Only real change was N:T271S in one of the two. (A29086T was inherited, but C->G muts like this are very rare.)
Reposted by Ryan Hisner

BA.3.2 still active in New Zealand.
🎃
🎃

Wastewater variants for week ended 9 Nov 2025:
💠NB.1.8.1 (Nimbus) 41.9%
💠XFG (Stratus) 23.0%
💠PE.1.4 17.2%
💠MC.10.2.1 9.6%
💠BA.3.2 3.7%
45% of NZ covered by testing:
💠Auckland
💠Rotorua
💠Wellington
💠Christchurch
💠Dunedin
💠Queenstown
poops.nz
2/
💠NB.1.8.1 (Nimbus) 41.9%
💠XFG (Stratus) 23.0%
💠PE.1.4 17.2%
💠MC.10.2.1 9.6%
💠BA.3.2 3.7%
45% of NZ covered by testing:
💠Auckland
💠Rotorua
💠Wellington
💠Christchurch
💠Dunedin
💠Queenstown
poops.nz
2/
ESR Wastewater Surveillance
poops.nz
November 27, 2025 at 12:02 PM
BA.3.2 still active in New Zealand.
🎃
🎃
Reposted by Ryan Hisner

Another US BA.3.2 wastewater detection.
This time from Bristol County, RI on Nov 11. It was about a third of the seqs.
I wonder if it was someone who flew back from Australia through SF?
@ryanhisner.bsky.social @snpoehlm.bsky.social @rajlabn.bsky.social
This time from Bristol County, RI on Nov 11. It was about a third of the seqs.
I wonder if it was someone who flew back from Australia through SF?
@ryanhisner.bsky.social @snpoehlm.bsky.social @rajlabn.bsky.social
November 26, 2025 at 11:01 PM
Another US BA.3.2 wastewater detection.
This time from Bristol County, RI on Nov 11. It was about a third of the seqs.
I wonder if it was someone who flew back from Australia through SF?
@ryanhisner.bsky.social @snpoehlm.bsky.social @rajlabn.bsky.social
This time from Bristol County, RI on Nov 11. It was about a third of the seqs.
I wonder if it was someone who flew back from Australia through SF?
@ryanhisner.bsky.social @snpoehlm.bsky.social @rajlabn.bsky.social
Reposted by Ryan Hisner

My work on SARS-CoV-2 variants informed a recent piece in The Lancet: "Epidemiological and virological update on the emerging SARS-CoV-2 variant BA.3.2".
The authors were kind enough to mention me and the Variant Hunters Ryan Hisner and Federico Gueli.
www.thelancet.com/journals/lan...
🧵
The authors were kind enough to mention me and the Variant Hunters Ryan Hisner and Federico Gueli.
www.thelancet.com/journals/lan...
🧵

Epidemiological and virological update on the emerging SARS-CoV-2 variant BA.3.2
The constant emergence of novel SARS-CoV-2 variants has driven the COVID-19 pandemic
and sustains the current endemic. Saltation variants, such as BA.2.86,1 encode highly
mutated spike (S) proteins th...
www.thelancet.com
November 25, 2025 at 1:30 AM
My work on SARS-CoV-2 variants informed a recent piece in The Lancet: "Epidemiological and virological update on the emerging SARS-CoV-2 variant BA.3.2".
The authors were kind enough to mention me and the Variant Hunters Ryan Hisner and Federico Gueli.
www.thelancet.com/journals/lan...
🧵
The authors were kind enough to mention me and the Variant Hunters Ryan Hisner and Federico Gueli.
www.thelancet.com/journals/lan...
🧵
Fascinating 🧵. The grotesquely mutated spike of this NJ Cryptic binds ACE2 very tightly.
It raises a broader question: Can cryptic wastewater-like lineages transmit?
YES
We knew it happened once. Now we know it's happened at least twice. The results in both cases were not pretty. 1/15
It raises a broader question: Can cryptic wastewater-like lineages transmit?
YES
We knew it happened once. Now we know it's happened at least twice. The results in both cases were not pretty. 1/15
This is wild.
Remember the NJ crytic lineage?
I posted 18 months ago that the Spike was too divergent to predict ACE2 binding, and asked if someone else could figure it out.
Some colleagues took me up on it.
Guess what they found?
1/
Remember the NJ crytic lineage?
I posted 18 months ago that the Spike was too divergent to predict ACE2 binding, and asked if someone else could figure it out.
Some colleagues took me up on it.
Guess what they found?
1/
November 22, 2025 at 7:19 PM
Fascinating 🧵. The grotesquely mutated spike of this NJ Cryptic binds ACE2 very tightly.
It raises a broader question: Can cryptic wastewater-like lineages transmit?
YES
We knew it happened once. Now we know it's happened at least twice. The results in both cases were not pretty. 1/15
It raises a broader question: Can cryptic wastewater-like lineages transmit?
YES
We knew it happened once. Now we know it's happened at least twice. The results in both cases were not pretty. 1/15
Reposted by Ryan Hisner

I think the number of downloads is not a good measure, because often I just quickly check something in the tree, I look for a certain mutation, a certain time and variant circulation, or take a screenshot of a tree for a talk. This was, and still is such an important resource!!
November 17, 2025 at 10:37 AM
I think the number of downloads is not a good measure, because often I just quickly check something in the tree, I look for a certain mutation, a certain time and variant circulation, or take a screenshot of a tree for a talk. This was, and still is such an important resource!!
Reposted by Ryan Hisner
Important read on challenges of continued SARS-CoV-2 surveillance provided by tools like Nextstrain, using GISAID data. Just one comment as someone who is using Nextstrain for my research, for keeping up to date and for scientific talks:
nextstrain.org/blog/2025-11...
nextstrain.org/blog/2025-11...

Nextstrain: Interruption to GISAID-based SARS-CoV-2 sequence analyses
Nextstrain blog post from 2025-11-06; author(s): Trevor Bedford, Richard Neher and the Nextstrain team
nextstrain.org
November 17, 2025 at 10:37 AM
Important read on challenges of continued SARS-CoV-2 surveillance provided by tools like Nextstrain, using GISAID data. Just one comment as someone who is using Nextstrain for my research, for keeping up to date and for scientific talks:
nextstrain.org/blog/2025-11...
nextstrain.org/blog/2025-11...
Excellent thread by @snpoehlm.bsky.social on BA.3.2, summarizing their recent study and outlining where we currently stand.
Two BA.3.2 sequences (of 67 total) showed up from New South Wales today, as BA.3.2 continues its creeping geographical spread.
Two BA.3.2 sequences (of 67 total) showed up from New South Wales today, as BA.3.2 continues its creeping geographical spread.
Our recent study on BA.3.2 is now available. Thanks to Markus Hoffmann, Lu Zhang and Georg Behrens for the great collaboration. Here’s a brief summary of our findings:
November 17, 2025 at 11:30 AM
Excellent thread by @snpoehlm.bsky.social on BA.3.2, summarizing their recent study and outlining where we currently stand.
Two BA.3.2 sequences (of 67 total) showed up from New South Wales today, as BA.3.2 continues its creeping geographical spread.
Two BA.3.2 sequences (of 67 total) showed up from New South Wales today, as BA.3.2 continues its creeping geographical spread.
Reposted by Ryan Hisner

Incredible story with echoes of frank abagnale jr. Apparently a guy pathologically obsessed with his reputation has established a record of building trust with researchers more understandably concerned with proper credit for their work, but the, uh, drawbacks of the arrangement remain unresolved.

November 16, 2025 at 6:59 PM
Incredible story with echoes of frank abagnale jr. Apparently a guy pathologically obsessed with his reputation has established a record of building trust with researchers more understandably concerned with proper credit for their work, but the, uh, drawbacks of the arrangement remain unresolved.
Reposted by Ryan Hisner
And for no reason... or rather no reason that can be stated publicly without revealing sordid ulterior motives—namely the monopolization of viral genomic data & analysis in the hands of Peter Bogner, a non-scientist & lifelong con artist who wields absolute power at GISAID. It's a tragedy & a farce.
November 16, 2025 at 6:56 PM
And for no reason... or rather no reason that can be stated publicly without revealing sordid ulterior motives—namely the monopolization of viral genomic data & analysis in the hands of Peter Bogner, a non-scientist & lifelong con artist who wields absolute power at GISAID. It's a tragedy & a farce.
Reposted by Ryan Hisner
I’ve used WA Health’s COVID-19 wastewater surveillance page to estimate the number of infections of BA.3.2.
I estimate ~300 BA.3.2.* infections in Perth for the latest week, and ~6,000 across the 12 weeks since BA.3.2.* was first detected.
#COVID19 #SARSCoV2 #BA_3_2 #Australia #WA #Perth
🧵
I estimate ~300 BA.3.2.* infections in Perth for the latest week, and ~6,000 across the 12 weeks since BA.3.2.* was first detected.
#COVID19 #SARSCoV2 #BA_3_2 #Australia #WA #Perth
🧵

November 15, 2025 at 1:29 AM
BA.3.2.2 nearly 50% of recent Western Australia sequences—perhaps on a path to dominance there?
In this latest batch, there's another furin-cleavage site (FCS)-adjacent ∆QT sequence. It's very closely related to the first one, so there's now no doubt that the deletion is real. 1/3
In this latest batch, there's another furin-cleavage site (FCS)-adjacent ∆QT sequence. It's very closely related to the first one, so there's now no doubt that the deletion is real. 1/3

November 13, 2025 at 12:23 AM
BA.3.2.2 nearly 50% of recent Western Australia sequences—perhaps on a path to dominance there?
In this latest batch, there's another furin-cleavage site (FCS)-adjacent ∆QT sequence. It's very closely related to the first one, so there's now no doubt that the deletion is real. 1/3
In this latest batch, there's another furin-cleavage site (FCS)-adjacent ∆QT sequence. It's very closely related to the first one, so there's now no doubt that the deletion is real. 1/3
Reposted by Ryan Hisner

1/ Pretty amazing new results showing an unexpected benefit of mRNA vaccines. Patients who received a COVID-19 mRNA vaccine within 100 days of starting immune checkpoint inhibitor (ICI) therapy survived almost twice as long. jenndowd.substack.com/p/can-mrna-v... #episky #medsky #cancer


November 10, 2025 at 12:12 PM
1/ Pretty amazing new results showing an unexpected benefit of mRNA vaccines. Patients who received a COVID-19 mRNA vaccine within 100 days of starting immune checkpoint inhibitor (ICI) therapy survived almost twice as long. jenndowd.substack.com/p/can-mrna-v... #episky #medsky #cancer
Reposted by Ryan Hisner

Excellent thread on BA.3 from @ryanhisner.bsky.social bsky.app/profile/ryan...
Do you remember BA.3—the weakling cousin of BA.1 & BA.2 that seemed to take the worst from each & had weaker ACE2 binding than even the ancestral Wuhan Virus?
After three years, BA.3 is back.
And it is transmitting.
Who saw this coming?
1/13
After three years, BA.3 is back.
And it is transmitting.
Who saw this coming?
1/13
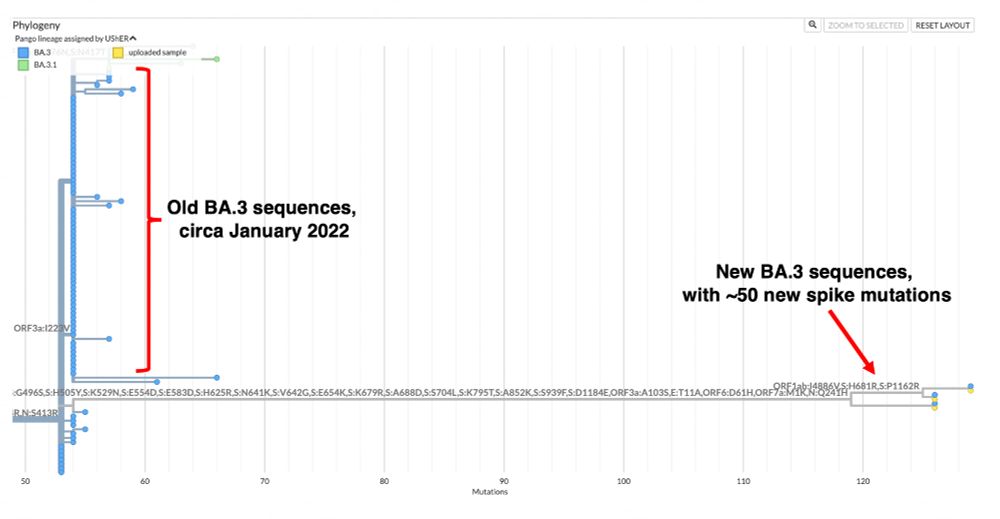
November 9, 2025 at 6:41 PM
Excellent thread on BA.3 from @ryanhisner.bsky.social bsky.app/profile/ryan...
Can't emphasize how damaging this is. A single non-scientist—Peter Bogner—holds all power & makes all decisions at GISAID & provides no justifications for any of them, except blatantly false ones.
He has that power because he conned rich & powerful people into giving it to him. Enough.
He has that power because he conned rich & powerful people into giving it to him. Enough.

I want to spell this out in case the implications aren't clear:
This means all public tools/webapps of GISAID data (all the ones you've been used to seeing thru the pandemic, as far as we can tell) are prohibited.
The file allowed this. Cut that - cut off all tools the public & others were using.
This means all public tools/webapps of GISAID data (all the ones you've been used to seeing thru the pandemic, as far as we can tell) are prohibited.
The file allowed this. Cut that - cut off all tools the public & others were using.

On Oct 1, 2025, GISAID informed us that they had ended updates to the flat file of SARS-CoV-2 genomic sequences and associated metadata that we had used to update Nextstrain analyses since Feb 2020. GISAID's stated rationale was that their "resources are limited". 1/5
November 10, 2025 at 2:46 AM
Can't emphasize how damaging this is. A single non-scientist—Peter Bogner—holds all power & makes all decisions at GISAID & provides no justifications for any of them, except blatantly false ones.
He has that power because he conned rich & powerful people into giving it to him. Enough.
He has that power because he conned rich & powerful people into giving it to him. Enough.
Reposted by Ryan Hisner
Continued surveillance and variant analysis is important, as highlighted by the emergence and spread of BA.3.2. Lack of data sharing by GISAID is bad news.

GISAID on providing data to @Nextstrain.org: "After consulting with our staff and advisors on the feasibility of keeping your global tree up-to-date, there was a clear consensus that continuing to generate, zip and move big files back and forth is not sustainable and a waste of resources."
🙃
🙃

Nextstrain: Interruption to GISAID-based SARS-CoV-2 sequence analyses
Nextstrain blog post from 2025-11-06; author(s): Trevor Bedford, Richard Neher and the Nextstrain team
next.nextstrain.org
November 6, 2025 at 10:14 PM
Continued surveillance and variant analysis is important, as highlighted by the emergence and spread of BA.3.2. Lack of data sharing by GISAID is bad news.
This is a total outrage and has crippled SARS-CoV-2 variant tracking and evolutionary analysis.
Updating a file and giving access to Nextstrain & Cov-Spectrum does not require extensive resources, so the official justification is a lie. There has to be an ulterior motive here.
Updating a file and giving access to Nextstrain & Cov-Spectrum does not require extensive resources, so the official justification is a lie. There has to be an ulterior motive here.
Nextstrain's daily-updated tree of SARS-CoV-2 genomes was my gateway into the world of viral phylogenetics in early 2020, and Nextstrain's beautiful interactive tree display is crucial to making usher.bio results usable. GISAID cutting off data harms global surveillance efforts. 🧵👇
On Oct 1, 2025, GISAID informed us that they had ended updates to the flat file of SARS-CoV-2 genomic sequences and associated metadata that we had used to update Nextstrain analyses since Feb 2020. GISAID's stated rationale was that their "resources are limited". 1/5
November 7, 2025 at 12:18 PM
This is a total outrage and has crippled SARS-CoV-2 variant tracking and evolutionary analysis.
Updating a file and giving access to Nextstrain & Cov-Spectrum does not require extensive resources, so the official justification is a lie. There has to be an ulterior motive here.
Updating a file and giving access to Nextstrain & Cov-Spectrum does not require extensive resources, so the official justification is a lie. There has to be an ulterior motive here.
BA.3.2 has arrived in the UK. One BA.3.2.2, collected October 5, was uploaded from Scotland today.
Same branch as recent BA.3.2.2 from Germany & Slovenia.
It has a few errors (S:ins214:ASDT is misread & ORF1a:E4388K is an artifact). Ignoring those, the one notable new mutation is N:N126K.
Same branch as recent BA.3.2.2 from Germany & Slovenia.
It has a few errors (S:ins214:ASDT is misread & ORF1a:E4388K is an artifact). Ignoring those, the one notable new mutation is N:N126K.

November 7, 2025 at 12:10 PM
BA.3.2 has arrived in the UK. One BA.3.2.2, collected October 5, was uploaded from Scotland today.
Same branch as recent BA.3.2.2 from Germany & Slovenia.
It has a few errors (S:ins214:ASDT is misread & ORF1a:E4388K is an artifact). Ignoring those, the one notable new mutation is N:N126K.
Same branch as recent BA.3.2.2 from Germany & Slovenia.
It has a few errors (S:ins214:ASDT is misread & ORF1a:E4388K is an artifact). Ignoring those, the one notable new mutation is N:N126K.
Reposted by Ryan Hisner
One of the greatest. I recently saw a car with a bumper sticker that said "Remember the Edmund Fitzgerald."
www.youtube.com/watch?v=FuzT...
www.youtube.com/watch?v=FuzT...

Gordon Lightfoot - Wreck Of The Edmund Fitzgerald (Official Audio)
YouTube video by Gordon Lightfoot
www.youtube.com
November 3, 2025 at 2:24 AM
One of the greatest. I recently saw a car with a bumper sticker that said "Remember the Edmund Fitzgerald."
www.youtube.com/watch?v=FuzT...
www.youtube.com/watch?v=FuzT...
Reposted by Ryan Hisner
BA.3.2 made it to New Zealand 👀
🦠Heads up. BA.3.2 has arrived in NZ
BA.3.2 detected in Aotearoa New Zealand wastewater
3.2% for the week to 5 October
BA.3.2 is not shown in the fortnight to 19 Oct. However, variants with a national percentage of < 1% are not included so BA.3.2 is likely to still be circulating
BA.3.2 detected in Aotearoa New Zealand wastewater
3.2% for the week to 5 October
BA.3.2 is not shown in the fortnight to 19 Oct. However, variants with a national percentage of < 1% are not included so BA.3.2 is likely to still be circulating

October 30, 2025 at 9:57 AM
BA.3.2 made it to New Zealand 👀
Reposted by Ryan Hisner
The cellular protease TMPRSS2 cleaves and activates the SARS-CoV-2 spike protein, priming it for membrane fusion and viral entry.
This step is essential for efficient infection of airway epithelial cells.
This step is essential for efficient infection of airway epithelial cells.
October 27, 2025 at 10:11 PM
The cellular protease TMPRSS2 cleaves and activates the SARS-CoV-2 spike protein, priming it for membrane fusion and viral entry.
This step is essential for efficient infection of airway epithelial cells.
This step is essential for efficient infection of airway epithelial cells.
First-world problem, I know, but I can't stand it when supplementary information is contained in 20 different files that have to be individually downloaded and viewed. Why not put it all in one PDF?

October 25, 2025 at 3:16 PM
First-world problem, I know, but I can't stand it when supplementary information is contained in 20 different files that have to be individually downloaded and viewed. Why not put it all in one PDF?
I can't reply to this comment, so I guess I'll quote-reply.
The most recent molnupiravir sequences have indeed been from Australia, with the MOV stats for the most recent being pictured below.
Once again, we see strong evidence that MOV-induced mutations are being positively selected. 1/4
The most recent molnupiravir sequences have indeed been from Australia, with the MOV stats for the most recent being pictured below.
Once again, we see strong evidence that MOV-induced mutations are being positively selected. 1/4

October 25, 2025 at 2:43 PM
I can't reply to this comment, so I guess I'll quote-reply.
The most recent molnupiravir sequences have indeed been from Australia, with the MOV stats for the most recent being pictured below.
Once again, we see strong evidence that MOV-induced mutations are being positively selected. 1/4
The most recent molnupiravir sequences have indeed been from Australia, with the MOV stats for the most recent being pictured below.
Once again, we see strong evidence that MOV-induced mutations are being positively selected. 1/4
BA.3.2.1 still hanging out in the Netherlands. Nothing new in spike in this latest one, though 3/4 nucleotide mutations are nonsynonymous (amino acid-changing) and all are in NSP3.

One more BA.3.2.1 from netherlands
cc @snpoehlm.bsky.social @ryanhisner.bsky.social @josetteschoenma.bsky.social ( i m apparently blocked by X, likely it doesn't like intl law)
cc @snpoehlm.bsky.social @ryanhisner.bsky.social @josetteschoenma.bsky.social ( i m apparently blocked by X, likely it doesn't like intl law)
October 23, 2025 at 4:39 PM
BA.3.2.1 still hanging out in the Netherlands. Nothing new in spike in this latest one, though 3/4 nucleotide mutations are nonsynonymous (amino acid-changing) and all are in NSP3.
I beg to differ! If it's not a sequencing mistake—and it looks clean—one of these BA.3.2 has something completely novel in SARS-CoV-2 evolution: an FCS-adjacent deletion!
One of the two QT repeats appears to have been deleted. I've never seen anything like this before. BA.3.2 is a different beast.
One of the two QT repeats appears to have been deleted. I've never seen anything like this before. BA.3.2 is a different beast.

October 22, 2025 at 11:46 AM
I beg to differ! If it's not a sequencing mistake—and it looks clean—one of these BA.3.2 has something completely novel in SARS-CoV-2 evolution: an FCS-adjacent deletion!
One of the two QT repeats appears to have been deleted. I've never seen anything like this before. BA.3.2 is a different beast.
One of the two QT repeats appears to have been deleted. I've never seen anything like this before. BA.3.2 is a different beast.