Jo Ma
@myzzzz.bsky.social
75 followers
460 following
12 posts
I believe innovation sparks from the intersection of difference
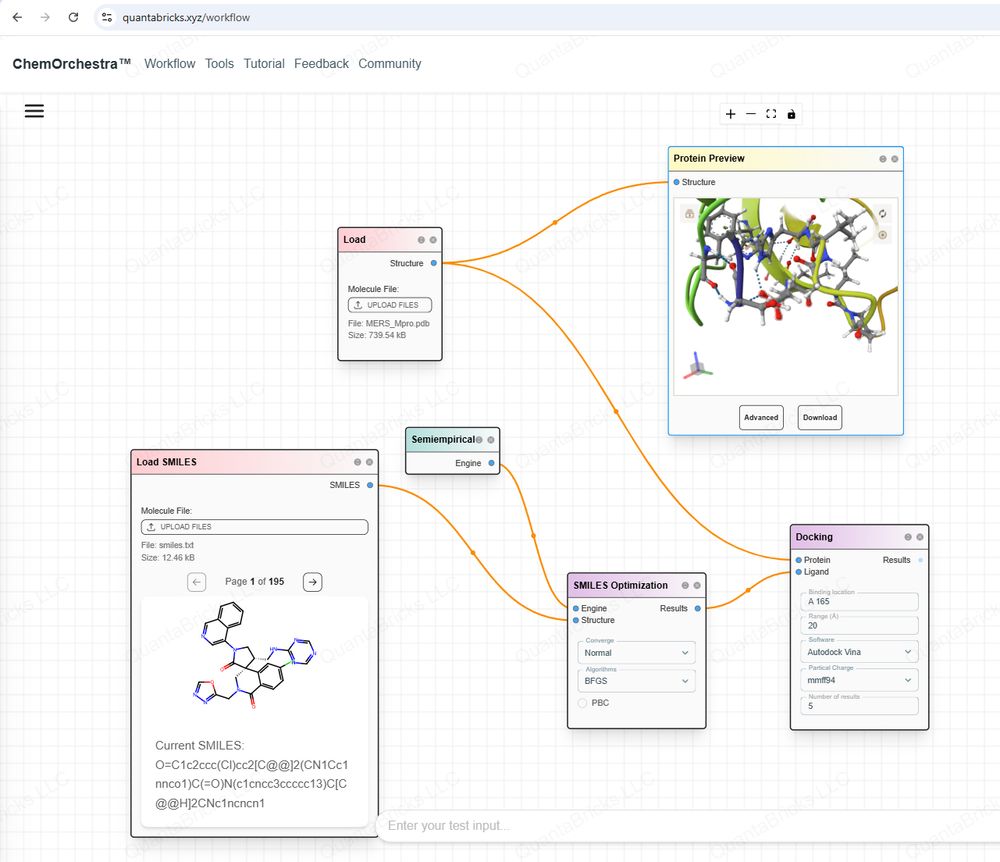
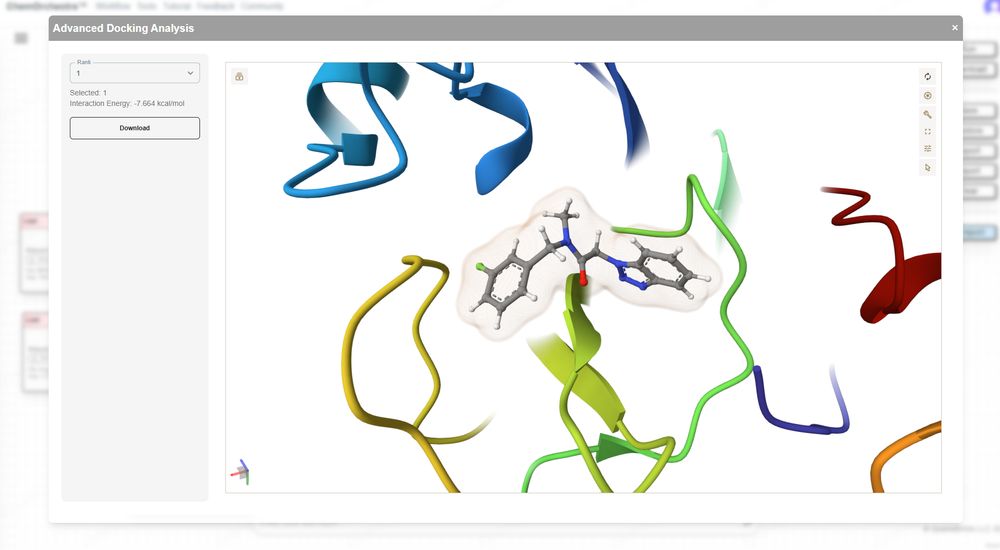
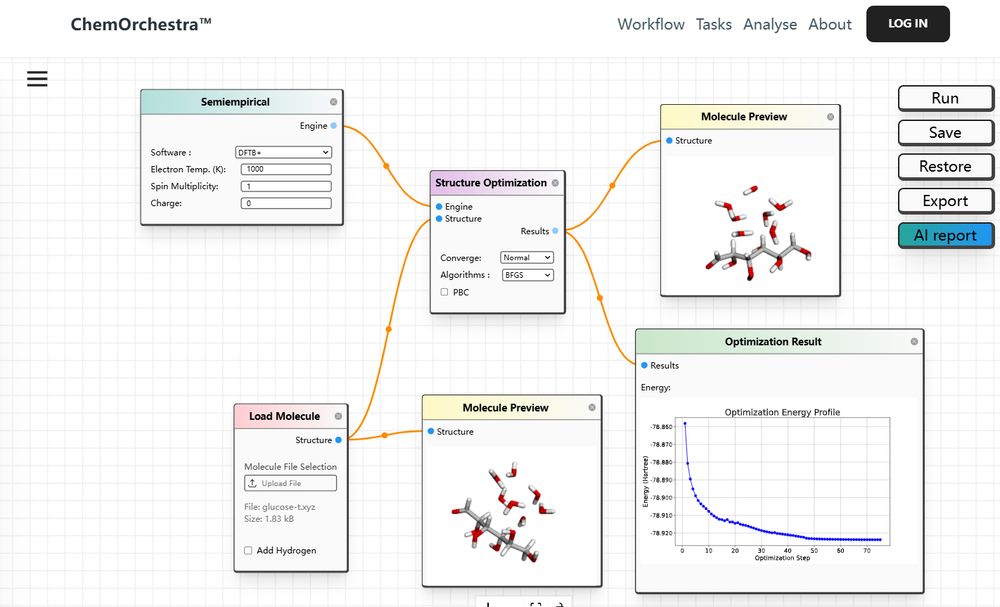
-Revolutionizing Drug & Materials Design for Everyone with Computational Chemistry https://quantabricks.substack.com/p/molecular-docking-in-a-easy-way
Posts
Media
Videos
Starter Packs
Jo Ma
@myzzzz.bsky.social
· 1d
Jo Ma
@myzzzz.bsky.social
· Jun 13
Jo Ma
@myzzzz.bsky.social
· Jun 13
Jo Ma
@myzzzz.bsky.social
· Jun 13
Jo Ma
@myzzzz.bsky.social
· Jun 13
Jo Ma
@myzzzz.bsky.social
· Jun 11
Jo Ma
@myzzzz.bsky.social
· Mar 21
Jo Ma
@myzzzz.bsky.social
· Mar 21