



Martin Fenk
@mfenk.bsky.social
210 followers
60 following
14 posts
PhD student @MPIIB Berlin | Exploring the captivating realm of microbial evolution within microbiomes #Microbiology #Evolution
Posts
Media
Videos
Starter Packs
Reposted by Martin Fenk

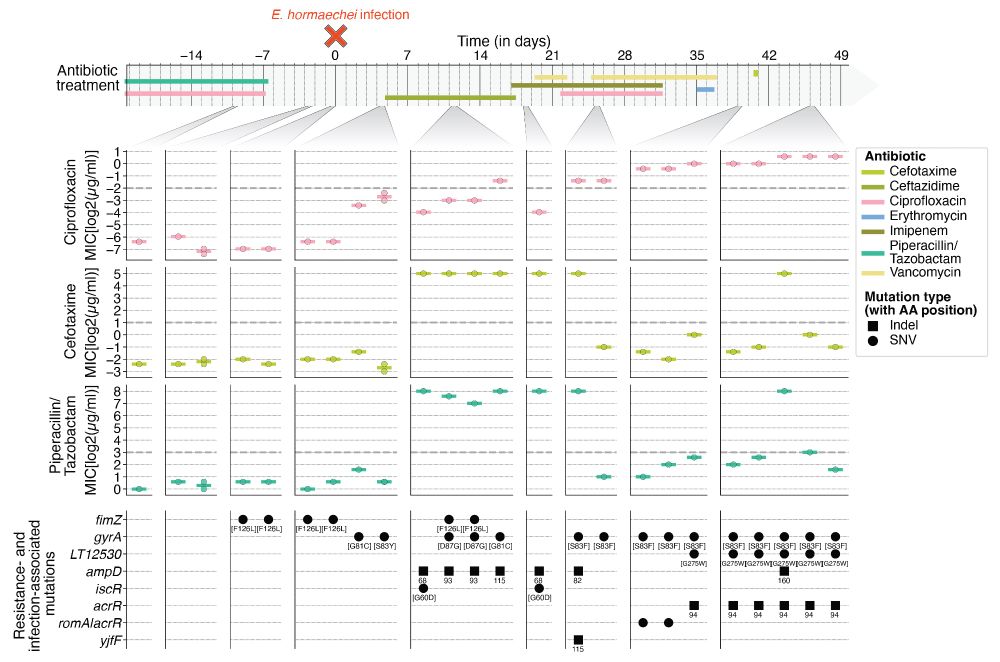
Is the human microbiome a source for hospital-associated infections (HAI) and are any genetic changes associated with HAI? In our new preprint, we longitudinally reconstruct the evolutionary processes within the human microbiome leading up to HAI: doi.org/10.1101/2025...

Colonization, translocation, and evolution of opportunistic pathogens during hospital-associated infections
Many commensal bacteria that peacefully reside in the human microbiome are also able to cause acute opportunistic infections. Emerging evidence suggests that within-host evolution contributes to infec...
doi.org








Reposted by Martin Fenk

Many zoonotic diseases are believed to have emerged during prehistory, but can we actually identify their past host range using ancient DNA? In the first publication of the Key Lab we present a 4000y old Yersinia pestis genome reconstructed from domesticated sheep. 🧵 www.biorxiv.org/content/10.1...

Bronze Age Yersinia pestis genome from sheep sheds light on hosts and evolution of a prehistoric plague lineage
Most human pathogens are of zoonotic origin. Many emerged during prehistory, coinciding with domestication providing more opportunities for spillover from original host species. However, we lack direc...
www.biorxiv.org