




Bloodsucking Parasites
@bloodsparasites.bsky.social
Science bulletin on bloodsucking arthropod vectors: flies, ticks, mosquitoes, mites, fleas, lice spreading dengue, malaria, WNV, Lyme, Rickettsia, Babesia, Bartonella -- MF
#Lyme disease control 2.0: Advances and opportunities coming with #Lyme disease vaccine VLA15 PLOSPathogens

#Lyme disease control 2.0: Advances and opportunities coming with #Lyme disease vaccine VLA15
by Ondrej Hajdusek, Kalvis Brangulis, Luise Robbertse, Rajesh Ghosh, Dino Di Carlo, Jan Perner
dlvr.it
December 16, 2025 at 2:47 AM
Differential antioxidant enzyme profiles reveal early molecular signatures of virulence in Trypanosoma cruzi DTU-TcI and DTU-TcVI strains PubMed

Differential antioxidant enzyme profiles reveal early molecular signatures of virulence in Trypanosoma cruzi DTU-TcI and DTU-TcVI strains
CONCLUSIONS: Enhanced early expression of antioxidant enzymes appears to correlate with T. cruzi virulence and persistence, suggesting a potential role for these molecules as biomarkers or therapeutic targets. These observations may help clarify strain-specific mechanisms contributing to Chagas disease pathogenesis.
dlvr.it
December 16, 2025 at 2:25 AM
Differential antioxidant enzyme profiles reveal early molecular signatures of virulence in Trypanosoma cruzi DTU-TcI and DTU-TcVI strains PubMed
Structural Dynamics and Allosteric Communication of a SARS-Like Bat Coronavirus Spike Glycoprotein bioRxivpreprint
Structural Dynamics and Allosteric Communication of a SARS-Like Bat Coronavirus Spike Glycoprotein
SARS-like bat coronaviruses (CoVs) pose ongoing public health risks due to their zoonotic potential, making it important to understand the molecular pathways driving their evolution. We recently showed that SHC014-CoV can infect human cell lines in an ACE2-dependent manner after acquiring two spike ectodomain mutations (F294L and A835D). However, how the wild-type (WT) SHC014 spike differs dynamically from these mutants remains unclear. Here, we built fully glycosylated ectodomain models of WT and three mutants (F294L, A835D, and the double mutant, DM) and performed triplicate all-atom molecular dynamics (MD) simulations for each variant. The two mutations exhibit epistasis, altering structural rearrangements relative to WT. Notably, the DM receptor binding domain (RBD) begins sampling the open conformation in our conventional MD. At the atomic level, the DM spike mitigates the dense negative packing introduced by A835D through a salt-bridge network, while F294L disrupts pi-mediated interactions, together enhancing RBD opening propensity which is critical for viral entry. Increased flexibility of the subdomain-2 620-loop further modulates DM RBD openness. Dynamical network analysis identified three allosteric communication pathways. In WT and F294L, Pathway 1 forms the baseline route linking the 620-loop to the RBD, whereas in A835D and DM it extends to the FPPR, reshaping long-range communication. Pathway 2 is conserved across variants but is most prominent in WT and F294L. Pathway 3 appears only in A835D and DM, compensating for reduced communication along Pathway 2. Overall, this work provides an atomistic perspective on SHC014 molecular adaptation during host-to-host transmission and highlights mechanistic features that may inform future therapeutic and pandemic-preparedness efforts.
dlvr.it
December 16, 2025 at 1:47 AM
Structural Dynamics and Allosteric Communication of a SARS-Like Bat Coronavirus Spike Glycoprotein bioRxivpreprint
Genetic characterization and identification of a recent discovered genotype of Theileria orientalis (Piroplasmida: Theileriidae) in water buffaloes (Bubalus bubalis) from the Amazon region, Brazil Ticks&TBD

Genetic characterization and identification of a recent discovered genotype of Theileria orientalis (Piroplasmida: Theileriidae) in water buffaloes (Bubalus bubalis) from the Amazon region, Brazil
Publication date: January 2026
Source: #ticks and #tick-borne Diseases, Volume 17, Issue 1
Author(s): João Paulo S. Alves, Pedro H.C. Rodrigues, Anisleidy P. Castillo, Antônio A. Fonseca Junior, Cairo H.S. de Oliveira, Rômulo C. Leite, José D. Barbosa, Júlia A.G. Silveira
dlvr.it
December 16, 2025 at 12:52 AM
Genetic characterization and identification of a recent discovered genotype of Theileria orientalis (Piroplasmida: Theileriidae) in water buffaloes (Bubalus bubalis) from the Amazon region, Brazil Ticks&TBD
Echolocation calls of some bat species in western Uganda bioRxivpreprint
Echolocation calls of some bat species in western Uganda
With the rise of accessible recording technology, passive acoustic monitoring can be an affordable and rapid way to assess species richness, even when individual animals cannot be captured due to regulatory or practical obstacles. Motivated by the relative lack of data and in partnership with the local populace, we recorded echolocation calls of freely-flying bats across six locations in rural western Uganda using opportunistic passive acoustic recordings. Frequency-modulated echolocation calls were recorded at all six locations, while constant-frequency calls were recorded only at sites near entrances to caves. Preliminary species identifications were made using Kaleidoscope Pro, habitat distribution maps for Uganda, and by reference to published work. We make our acoustic recordings publicly available to serve as a resource for further explorations of the richness of bat species in Uganda.
dlvr.it
December 16, 2025 at 12:13 AM
Echolocation calls of some bat species in western Uganda bioRxivpreprint
A Tetrasaccharide Containing a Galactofuranosyl Determinant of Mucins from Trypanosoma cruzi Inhibits the Parasite Adhesion on Triatoma infestans Rectum PubMed_
A Tetrasaccharide Containing a Galactofuranosyl Determinant of Mucins from Trypanosoma cruzi Inhibits the Parasite Adhesion on Triatoma infestans Rectum
Trypanosoma cruzi, the etiologic agent of Chagas disease, is a protozoan parasite transmitted to humans via the feces of blood-sucking triatomine vectors such as Triatoma infestans. It has been shown that galactofuranose-containing oligosaccharides present in mucins of T. cruzi are involved in parasite adhesion to the T. infestans rectal ampoule, a key step in the differentiation programme from dividing epimastigote to nondividing mammal-infective metacyclic forms of the parasite. To further...
dlvr.it
December 15, 2025 at 9:26 PM
A Tetrasaccharide Containing a Galactofuranosyl Determinant of Mucins from Trypanosoma cruzi Inhibits the Parasite Adhesion on Triatoma infestans Rectum PubMed_
Acceptability and preferences for dual-active ingredient long-lasting insecticidal nets in rural Tanzania: a mixed-methods study PubMed
Acceptability and preferences for dual-active ingredient long-lasting insecticidal nets in rural Tanzania: a mixed-methods study
CONCLUSION: Acceptability and sustained use of dual-AI LLINs are shaped by perceived efficacy, comfort, and net integrity, while barriers like bed bugs and skin irritation reduce use. Addressing non-target pest issues, targeting different groups of users with tailored education, and integrating user perception into LLIN procurement can enhance uptake and impact. It is recommended that manufacturers and policymakers consider these community-informed insights to guide the development and...
dlvr.it
December 15, 2025 at 8:29 PM
Acceptability and preferences for dual-active ingredient long-lasting insecticidal nets in rural Tanzania: a mixed-methods study PubMed
Ferritin-mediated regulation of gut microbiota homeostasis promotes blood-feeding adaptation in the #tick Haemaphysalis doenitzi PubMed_
Ferritin-mediated regulation of gut microbiota homeostasis promotes blood-feeding adaptation in the #tick Haemaphysalis doenitzi
As obligate hematophagous parasites, ticks have evolved to cope with substantial amounts of iron and exogenous microorganisms present in host blood during feeding. In ticks, ferritin plays an important role in maintaining the oxidative balance of gut and the homeostasis of the microbial community structure, but its regulatory mechanism has not yet been clarified. This study successfully identified a ferritin gene from Haemaphysalis doenitzi, named Hd-fer, and further studied the function of...
dlvr.it
December 15, 2025 at 4:36 PM
Ferritin-mediated regulation of gut microbiota homeostasis promotes blood-feeding adaptation in the #tick Haemaphysalis doenitzi PubMed_
Evidence for Transcription and Horizontal Gene Transfer in Dipteran Germline-Restricted Chromosomes bioRxivpreprint
Evidence for Transcription and Horizontal Gene Transfer in Dipteran Germline-Restricted Chromosomes
Germline-restricted chromosomes (GRCs) constitute a unique class of chromosomes confined to reproductive cells. Arising across multiple, evolutionarily distant lineages, GRCs have been identified in three insect families (non-biting midges, gall midges, and fungus gnats), each within the order Diptera. Genomic characterisation in fungus gnats has revealed GRCs to be large, gene-rich chromosomes, which, together with their persistence over millennia, implies they play important biological roles within this clade. However, transcription from these chromosomes has yet to be demonstrated, leaving key questions about their function and activity unresolved. Here, we provide the first direct evidence of GRC-linked gene expression in the fungus gnat Bradysia coprophila, integrating RNA-seq data with cytological observations across multiple developmental stages. We report that GRCs express functional genes, though overall transcription is highly limited, likely due to biological factors, such as transcriptional silencing in specific germline cell types, and technical constraints, including filtering to avoid mismapping from core chromosome paralogues. We identify 15 confidently expressed GRC-linked genes using stringent criteria, including five insect homologues of unknown function and nine resembling transposable elements, and report horizontal acquisition of a ~290 kb bacterial-derived region on GRC2. Furthermore, we perform in vitro immunofluorescence staining and confocal microscopy, which indicate increased GRC activity in female oocytes. Overall, these findings establish GRCs in fungus gnats as transcriptionally active, albeit tightly regulated and highly dynamic, chromosomes capable of expressing both endogenous and potentially horizontally acquired genes.
dlvr.it
December 15, 2025 at 4:34 PM
Evidence for Transcription and Horizontal Gene Transfer in Dipteran Germline-Restricted Chromosomes bioRxivpreprint
Molecular characterization of Hepatozoon spp. in cats living in Germany and other European countries PubMed
Molecular characterization of Hepatozoon spp. in cats living in Germany and other European countries
Hepatozoon spp. are increasingly reported in cats from Mediterranean countries, but data for Central and Northern Europe remain limited. This study investigated the occurrence and molecular diversity of Hepatozoon spp. in 1357 blood samples from cats living in Germany and other European countries using real-time PCR targeting the 18S rRNA gene. Hepatozoon spp. DNA was detected in 58 cats (4.3 %; 95 %-CI: 3.3-5.5 %). Thirty-seven positive samples were further analyzed by conventional PCR and...
dlvr.it
December 15, 2025 at 2:32 PM
Molecular characterization of Hepatozoon spp. in cats living in Germany and other European countries PubMed
Pathogens, Vol. 14, Pages 1286: Unraveling the Diversity of Haemosporidians in Brazilian Non-Passerine Birds: Insights from Midwestern Brazil Pathogens
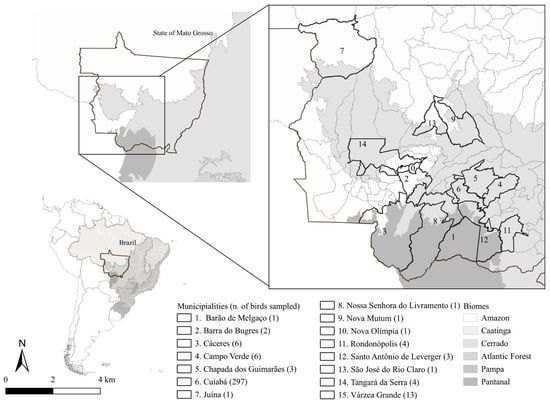
Pathogens, Vol. 14, Pages 1286: Unraveling the Diversity of Haemosporidians in Brazilian Non-Passerine Birds: Insights from Midwestern Brazil
Avian haemosporidians have been widely studied because they provide important insights into parasite distribution and diversity. However, most available data come from passerines, resulting in gaps regarding other bird groups, primarily due to the difficulty of sampling non-passerines in natural environments. Thus, we aimed to detect infections caused by Plasmodium spp. and Haemoproteus spp. through molecular and morphological analyses of blood samples from non-passerine birds in Midwestern Brazil. We evaluated 344 individuals from 60 species across 16 non-passerine orders. Among them, 18.89% (n = 65) were infected with haemosporidians. Molecular analyses identified four Plasmodium species: P. nucleophilum, which was detected in a broad range of host species; P. juxtanucleare, detected in Gallus gallus; P. paranucleophilum, found to infect Rupornis magnirostris; and P. elongatum in Mustelirallus albicollis. Additionally, an undescribed Plasmodium lineage was detected in Nycticorax nycticorax. We also identified four new Haemoproteus lineages infecting Patagioenas picazuro, Asio clamator, Athene cunicularia, and Tyto furcata. Additionally, the haplotype previously described in Mycteria americana was detected once more in this host. By revealing new lineages and expanding knowledge of parasite biodiversity, this study underscores the importance of non-passerine hosts and the need for further research on their evolutionary and host–parasite relationships.
dlvr.it
December 15, 2025 at 10:11 AM
Pathogens, Vol. 14, Pages 1286: Unraveling the Diversity of Haemosporidians in Brazilian Non-Passerine Birds: Insights from Midwestern Brazil Pathogens
The dynamic of #Trypanosoma cruzi transmissibility in field-caught Mepraia spinolai (Hemiptera: Reduviidae) of contrasting seasons: a developmental stage-dependent study #ActaTropica

The dynamic of #Trypanosoma cruzi transmissibility in field-caught Mepraia spinolai (Hemiptera: Reduviidae) of contrasting seasons: a developmental stage-dependent study
Publication date: Available online 14 December 2025
Source: Acta Tropica
Author(s): Nicol Quiroga, Francisca Farías, Angélica López, Carezza Botto-Mahan, Aldo Solari
dlvr.it
December 15, 2025 at 8:49 AM
The dynamic of #Trypanosoma cruzi transmissibility in field-caught Mepraia spinolai (Hemiptera: Reduviidae) of contrasting seasons: a developmental stage-dependent study #ActaTropica
Asymptomatic Plasmodium infection and predictors among schoolchildren in Bahir Dar Zuria District, Northwest Ethiopia PubMed
Asymptomatic Plasmodium infection and predictors among schoolchildren in Bahir Dar Zuria District, Northwest Ethiopia
CONCLUSION: This study revealed that asymptomatic schoolchildren serve as significant reservoirs for malaria, challenging elimination efforts. The findings emphasize the need for integrated interventions such as active case detection and treatment, improved long-lasting insecticidal net coverage, and environmental management. Importantly, comprehensive surveillance and seasonal malaria chemoprevention are also recommended to reduce silent transmission and support targeted control strategies.
dlvr.it
December 15, 2025 at 8:35 AM
Asymptomatic Plasmodium infection and predictors among schoolchildren in Bahir Dar Zuria District, Northwest Ethiopia PubMed
Microfluidic Immunocapture Device for Direct Detection of Lyme Disease bioRxivpreprint
Microfluidic Immunocapture Device for Direct Detection of Lyme Disease
Lyme Disease is a multisystem infectious disease caused by the Borellia burgdorferi complex, and is a growing threat to public health. Approximately 476,000 people are infected with Lyme in the United States each year. Although Lyme is readily treated with antibiotics when detected early, early detection remains difficult. Current testing remains difficult because the standard 2-tiered ELISA/Western assay indirectly detects Lyme via measurement of a host immune response, which suffers from an inherent time-lag in host antibody production. A direct test for Lyme Disease would overcome these inherent limitations. To this end we report on the first microfluidic immunocapture device for Lyme Disease. We engineered a geometrically enhanced differential immunocapture (GEDI) technology to capture whole-organism Borrelia for direct on-chip detection. This approach is potentially amenable with other work in the field to develop direct PCR or aptamer tests for Lyme Disease, as our device could serve as a platform to drastically enhance the concentration of present Borrelia into a small volume.
dlvr.it
December 15, 2025 at 2:50 AM
Microfluidic Immunocapture Device for Direct Detection of Lyme Disease bioRxivpreprint
Oviposition response of Aedes mosquitoes to different cement types: a field-based study in urban Sri Lanka PubMed
Oviposition response of Aedes mosquitoes to different cement types: a field-based study in urban Sri Lanka
CONCLUSION: The BHC and PCC cement types significantly deterred Aedes oviposition compared to other substrates, indicating that construction materials can influence mosquito breeding behavior.
dlvr.it
December 15, 2025 at 2:37 AM
Oviposition response of Aedes mosquitoes to different cement types: a field-based study in urban Sri Lanka PubMed
Genomic Flexibility Through Extrachromosomal Amplifications: A Leishmania Survival Strategy bioRxivpreprint
Genomic Flexibility Through Extrachromosomal Amplifications: A Leishmania Survival Strategy
Leishmania parasites modulate gene copy number through extrachromosomal DNA (ecDNA) amplification, enabling adaptation to environmental stress. Under drug pressure, both linear and circular ecDNA amplifications (amplicons) carrying resistance genes emerge. However, how these ecDNA structures form, diversify, and coexist remains poorly understood. Here, using experimental evolution and Oxford Nanopore long-read sequencing, we show that a single clonal population of drug resistant Leishmania produces a variety of linear and circular amplicons. As antimonial pressure increases, linear amplicons transition into circular forms, with high-stress conditions favoring circular amplicons carrying at least two copies of the resistance gene. Using the Nanopore long reads, we map recombination events driving linear and circular amplicon formation. Our model suggests that gene duplication in the amplicons originates from inter-chromatid homologous recombination, leading to an intermediate intra-chromosomal duplication, followed by a second homologous recombination event. Additionally, different Leishmania species exhibited distinct biases toward linear or circular amplification under identical drug conditions, suggesting species-specific adaptive strategies. Together, these findings define recombination-driven ecDNA dynamics as a central axis of genomic plasticity in Leishmania and underscore the potential for targeting ecDNA in therapeutic and diagnostic strategies against Leishmania and related pathogens.
dlvr.it
December 15, 2025 at 1:16 AM
Genomic Flexibility Through Extrachromosomal Amplifications: A Leishmania Survival Strategy bioRxivpreprint
Pathogens, Vol. 14, Pages 1282: Importance of Different Parameters for Monitoring Dogs with Leishmania infantum Infections in a Non-Endemic Country Pathogens

Pathogens, Vol. 14, Pages 1282: Importance of Different Parameters for Monitoring Dogs with Leishmania infantum Infections in a Non-Endemic Country
Leishmania (L.) infantum infections in dogs can cause severe recurrent disease. The aim of this study was to investigate different parameters for early detection of disease relapses in L. infantum-infected dogs in Germany. Fifty-two dogs naturally infected with L. infantum were enrolled. During the one-year study period, all dogs remained outside of endemic areas and attended study appointments every three months, including physical examination, blood pressure measurement, complete blood count with differential, serum biochemistry with symmetrical dimethylarginine and C-reactive protein, complete urinalysis including urine protein-to-creatinine ratio, L. infantum PCR, and antibody ELISA. Disease relapse was defined as deterioration of clinical or laboratory parameters in dogs that had achieved complete or partial remission before. Univariable and multivariable Bayesian logistic regression were used to identify predictors of disease relapse. Lymphadenopathy (p < 0.01; OR = 6.93), seborrhea/hypotrichosis (p = 0.02; OR = 8.02), and proteinuria (p < 0.01; OR = 9.14) were significantly associated with upcoming disease relapses (n = 10; 9/52 dogs), while associations between higher antibody levels and upcoming disease relapses trended towards significance (p = 0.06; OR = 1.03). Different parameters are important for an early diagnosis of disease relapse in canine leishmaniosis and should thus be regularly assessed and interpreted accordingly in the monitoring of L. infantum-infected dogs.
dlvr.it
December 14, 2025 at 10:13 PM
Pathogens, Vol. 14, Pages 1282: Importance of Different Parameters for Monitoring Dogs with Leishmania infantum Infections in a Non-Endemic Country Pathogens
Susceptibility to the larvicide pyriproxyfen and genotyping of kdr mutations in two #Aedes aegypti populations from Maranhão State, Brazil #ActaTropica

Susceptibility to the larvicide pyriproxyfen and genotyping of kdr mutations in two #Aedes aegypti populations from Maranhão State, Brazil
Publication date: Available online 13 December 2025
Source: Acta Tropica
Author(s): Valéria Cristina Soares Pinheiro, Juliete Lima Viana, Joelma Soares da Silva, Márcia Verônica Pereira Gonçalves, Aylane Tamara dos Santos Andrade, Ademir Jesus Martins
dlvr.it
December 14, 2025 at 8:52 PM
Susceptibility to the larvicide pyriproxyfen and genotyping of kdr mutations in two #Aedes aegypti populations from Maranhão State, Brazil #ActaTropica
Socioeconomic vulnerability and the management of domestic animal hosts in urban environments: a one health issue PubMed
Socioeconomic vulnerability and the management of domestic animal hosts in urban environments: a one health issue
CONCLUSIONS: This study highlights the need for basic animal health measures, such as sterilization, improved nutrition, deworming, and controlling street access, to reduce the competence of these animals as hosts of infectious agents, considering the vulnerability of these communities. Therefore, it is necessary to expand public policies focused on the promotion and prevention of comprehensive health, extending these measures to animal health.
dlvr.it
December 14, 2025 at 8:40 PM
Socioeconomic vulnerability and the management of domestic animal hosts in urban environments: a one health issue PubMed
Molecular characterization of Hepatozoon spp. in #cats living in Germany and other European countries Ticks&TBD

Molecular characterization of Hepatozoon spp. in #cats living in Germany and other European countries
Publication date: January 2026
Source: #ticks and #tick-borne Diseases, Volume 17, Issue 1
Author(s): Vera Geisen, Nikola Pantchev, Yury Zablotski, Majda Globokar Vrhovec, Katrin Hartmann, Michéle Bergmann, Gastón Moré, Walter Basso
dlvr.it
December 14, 2025 at 8:09 PM
Molecular characterization of Hepatozoon spp. in #cats living in Germany and other European countries Ticks&TBD
Complete genome sequence and genetic features of a novel Pseudomonas sp. isolate (CAM1A) from tsetse fly gut captured in Dodeo, Cameroon PubMed
Complete genome sequence and genetic features of a novel Pseudomonas sp. isolate (CAM1A) from tsetse fly gut captured in Dodeo, Cameroon
No abstract
dlvr.it
December 14, 2025 at 2:43 PM
Complete genome sequence and genetic features of a novel Pseudomonas sp. isolate (CAM1A) from tsetse fly gut captured in Dodeo, Cameroon PubMed
Pathogens, Vol. 14, Pages 1281: Interlaboratory Concordance of a Multiplex ELISA for Lyme and Lyme-like Illness Using Australian Samples and Commercial Reference Panels: A Proof-of-Concept Study Pathogens

Pathogens, Vol. 14, Pages 1281: Interlaboratory Concordance of a Multiplex ELISA for Lyme and Lyme-like Illness Using Australian Samples and Commercial Reference Panels: A Proof-of-Concept Study
Tick bites acquired in the northern or southern hemisphere can transmit microbes that may cause illness. The most prevalent infection is Lyme borreliosis (LB), with all proven cases to date having been acquired in the northern hemisphere. The existence of endemic LB in Australia has not been proven explicitly, and there is uncertainty concerning the cause of “Lyme-like” disease (LLD) in Australia. As many tick-borne diseases (TBDs) are diagnosed by serology, validated assays for use in both the northern and southern hemispheres are required. Using a multiplex enzyme-linked immunosorbent assay (TICKPLEX®), two independent laboratories tested a total of 53 well-characterized reference sera that consisted of 33 samples from northern hemisphere patients with confirmed tick-borne disease (TBD) and 20 randomly selected sera from Australian patients with suspected TBDs, presenting with or without LLD. Antibody responses to multiple microbial antigens from causative agents of TBDs were found. High concordance between laboratories was demonstrated on this small set of samples. The results obtained provide the basis for further evaluation of TICKPLEX® on a larger number of samples from Australian patients with suspected TBDs. These findings should be considered preliminary, providing proof-of-concept evidence that warrants validation in larger, clinically diverse cohorts.
dlvr.it
December 14, 2025 at 10:16 AM
Pathogens, Vol. 14, Pages 1281: Interlaboratory Concordance of a Multiplex ELISA for Lyme and Lyme-like Illness Using Australian Samples and Commercial Reference Panels: A Proof-of-Concept Study Pathogens
Liquid-liquid phase separation (LLPS) as a sensing and adaptation mechanism: An evidence-based hypothesis on AP2 transcription factors in the malaria parasite bioRxivpreprint
Liquid-liquid phase separation (LLPS) as a sensing and adaptation mechanism: An evidence-based hypothesis on AP2 transcription factors in the malaria parasite
Background: Protein liquid-liquid phase separation (LLPS) can be driven by prion-like domains (PrLDs) inside intrinsically disordered regions (IDRs). The causing agent of the deadliest form of human malaria, Plasmodium falciparum, has abundant prion-like proteins whose aggregation is presumed to have a functional role. Multiple members of the largest family of transcription factors in P. falciparum, AP2 (PfAP2), responsible for adapting the parasite gene response in different scenarios, were found in aggregation-prone protein screenings. Results: We show that the PfAP2s members carry the physicochemical determinants to perform LLPS forming biomolecular condensates in vivo. The long IDRs of PfAP2s could sense changes in the cellular microenvironment, and their PrLDs could drive conformational rearrangements. PfAP2s do not function as centralizing hubs for protein-protein interaction networks, but display significant preferred interactions among themselves, establishing a large, connected subnetwork. Predictions suggest that all PfAP2s have regions to localize into LLPS-condensates, while larger PfAP2s could initiate condensation. We show that four PfAP2 members co-localize in live P. falciparum, bearing the potential to be engaged in LLPS-condensates. Conclusion: We present bioinformatics analyses and experimental data obtained in live parasites suggesting that PfAP2s are able to direct LLPS in P. falciparum. We propose a model in which sensing by the parasite of cellular stresses like host transfer, temperature changes and energy depletion, and the corresponding gene responses are driven by LLPS where the PfAP2 family plays a fundamental role. Finally, we postulate targeting PfAP2 as a new therapeutic antimalarial strategy to curb the emergence of drug-resistant parasites.
dlvr.it
December 14, 2025 at 9:56 AM
Liquid-liquid phase separation (LLPS) as a sensing and adaptation mechanism: An evidence-based hypothesis on AP2 transcription factors in the malaria parasite bioRxivpreprint
Single-cell multiomics reveals epigenetic rewiring of splenic memory B cells during murine malaria reinfection bioRxivpreprint
Single-cell multiomics reveals epigenetic rewiring of splenic memory B cells during murine malaria reinfection
Malaria induces slow, gradually acquired, non-sterilizing immunity whose cellular and regulatory underpinnings remain incompletely understood. Here, we combine a sequential Plasmodium yoelii 17XNL infection model in BALB/c mice, in which primary parasitemia resolves spontaneously and confers robust protection upon homologous reinfection, with single-cell RNA and chromatin accessibility profiling to dissect how primary infection and recall reshape splenic immunity, with a focus on B cells. We generate a multiomic atlas of >50,000 splenic mononuclear cells, resolving thirteen major immune lineages and 48 subpopulations, and show that B cells dominate the response and diversify into naive/mature, germinal center, memory, and plasmablast compartments. Trajectory analysis reveals distinct differentiation paths towards germinal center, memory, and mature B cells, and uncovers infection-dependent shifts in transcription factor activity, cis-regulatory element usage, and gene regulatory networks. Reinfection is associated with a shift in memory B-cell composition and transcriptional programs towards extrafollicular-like, IgM-conventional memory B cells together with epigenetic modules linked to rapid antibody production. Together, these data provide a systems-level view of B cell plasticity in experimental malaria and provides a mechanistic framework from a highly protective P. yoelii reinfection model with implications for understanding non-sterilizing immunity in endemic settings.
dlvr.it
December 14, 2025 at 9:43 AM
Single-cell multiomics reveals epigenetic rewiring of splenic memory B cells during murine malaria reinfection bioRxivpreprint