



Michael Hall
@mbhall88.bsky.social
Bioinformatics geek 🤓 crafting Rust-y tools 🦀 for microbial genomes 🦠 🧬.
Trying to master Dad mode 👨🍼
See what I'm up to here: https://github.com/mbhall88
Trying to master Dad mode 👨🍼
See what I'm up to here: https://github.com/mbhall88
So nohuman now ships an unmasked HPRC.r2 DB by default, with optional dataset selection.
If you’ve used nohuman before, I highly recommend updating to v0.5.0 and re-downloading the new DB.
Repo: github.com/mbhall88/nohuman
Keep your metagenomes clean 🧹🧬
If you’ve used nohuman before, I highly recommend updating to v0.5.0 and re-downloading the new DB.
Repo: github.com/mbhall88/nohuman
Keep your metagenomes clean 🧹🧬

GitHub - mbhall88/nohuman: Remove human reads from a sequencing run
Remove human reads from a sequencing run. Contribute to mbhall88/nohuman development by creating an account on GitHub.
github.com
November 20, 2025 at 6:50 AM
So nohuman now ships an unmasked HPRC.r2 DB by default, with optional dataset selection.
If you’ve used nohuman before, I highly recommend updating to v0.5.0 and re-downloading the new DB.
Repo: github.com/mbhall88/nohuman
Keep your metagenomes clean 🧹🧬
If you’ve used nohuman before, I highly recommend updating to v0.5.0 and re-downloading the new DB.
Repo: github.com/mbhall88/nohuman
Keep your metagenomes clean 🧹🧬
At the same time, I realised the Human Pnagenome Reference Consortium had made a second release of genomes.
So I rebuilt release 1 without masking, and added a release 2 database with no masking. The improvement in detection accuracy was substantial:
So I rebuilt release 1 without masking, and added a release 2 database with no masking. The improvement in detection accuracy was substantial:

November 20, 2025 at 6:50 AM
At the same time, I realised the Human Pnagenome Reference Consortium had made a second release of genomes.
So I rebuilt release 1 without masking, and added a release 2 database with no masking. The improvement in detection accuracy was substantial:
So I rebuilt release 1 without masking, and added a release 2 database with no masking. The improvement in detection accuracy was substantial:
Stars are level of p value (description is in the figure caption in the paper)
November 7, 2025 at 7:43 PM
Stars are level of p value (description is in the figure caption in the paper)
True.
Thanks for the great questions and discussion
Thanks for the great questions and discussion
November 7, 2025 at 11:07 AM
True.
Thanks for the great questions and discussion
Thanks for the great questions and discussion
Correct. Yeah I guess mash on a random subset should perform similarly. Haven’t looked at that though.
November 7, 2025 at 11:05 AM
Correct. Yeah I guess mash on a random subset should perform similarly. Haven’t looked at that though.
It’s a decent sample size at 3000. But I guess more would always be better. I wanted to use refseq genomes which has long read data to be as sure as possible about the true size
There is likely inherent biases though based on error rates in reads for the kmer based methods
There is likely inherent biases though based on error rates in reads for the kmer based methods
November 7, 2025 at 11:04 AM
It’s a decent sample size at 3000. But I guess more would always be better. I wanted to use refseq genomes which has long read data to be as sure as possible about the true size
There is likely inherent biases though based on error rates in reads for the kmer based methods
There is likely inherent biases though based on error rates in reads for the kmer based methods
- Overlaps are pairwise alignment with minima2 (FFI)
-Thanks!
- See other thread where I have answered this
-Thanks!
- See other thread where I have answered this
November 7, 2025 at 11:01 AM
- Overlaps are pairwise alignment with minima2 (FFI)
-Thanks!
- See other thread where I have answered this
-Thanks!
- See other thread where I have answered this
I just used mash v2.3. The supplement has an exploration of the best parameters to use for mash to estimate genome size. Mash was the fastest tool though.
November 7, 2025 at 10:58 AM
I just used mash v2.3. The supplement has an exploration of the best parameters to use for mash to estimate genome size. Mash was the fastest tool though.
Thanks for appreciating the plots. I obsessed a lot over them. I created a repo for the colour palette too if you’re interested in that github.com/mbhall88/cud

GitHub - mbhall88/cud: Color Universal Design colourblind-friendly python matplotlib palette
Color Universal Design colourblind-friendly python matplotlib palette - mbhall88/cud
github.com
November 7, 2025 at 10:56 AM
Thanks for appreciating the plots. I obsessed a lot over them. I created a repo for the colour palette too if you’re interested in that github.com/mbhall88/cud
the bars are pair wise statistical comparisons. I only show the significant ones so as not to over clutter the plot
November 7, 2025 at 10:52 AM
the bars are pair wise statistical comparisons. I only show the significant ones so as not to over clutter the plot
And lastly, a HUGE thank you to @lachlanjmc.bsky.social for a lot of the methodological heavy lifting when we were coming up with the idea
November 7, 2025 at 3:21 AM
And lastly, a HUGE thank you to @lachlanjmc.bsky.social for a lot of the methodological heavy lifting when we were coming up with the idea
Try LRGE here: github.com/mbhall88/lrge
(installable from wherever you get your podcasts 😉)
(installable from wherever you get your podcasts 😉)

GitHub - mbhall88/lrge: Genome size estimation from long read overlaps
Genome size estimation from long read overlaps. Contribute to mbhall88/lrge development by creating an account on GitHub.
github.com
November 7, 2025 at 3:19 AM
Try LRGE here: github.com/mbhall88/lrge
(installable from wherever you get your podcasts 😉)
(installable from wherever you get your podcasts 😉)
You might remember the preprint from late last year... Reviews/Publication were delayed while I was on parental leave. We extended validation to include H. sapiens, which lead to smarter handling of contained overlaps in repetitive genomes. Big shout-out to Chenxi Zhou for leading that part
November 7, 2025 at 3:19 AM
You might remember the preprint from late last year... Reviews/Publication were delayed while I was on parental leave. We extended validation to include H. sapiens, which lead to smarter handling of contained overlaps in repetitive genomes. Big shout-out to Chenxi Zhou for leading that part
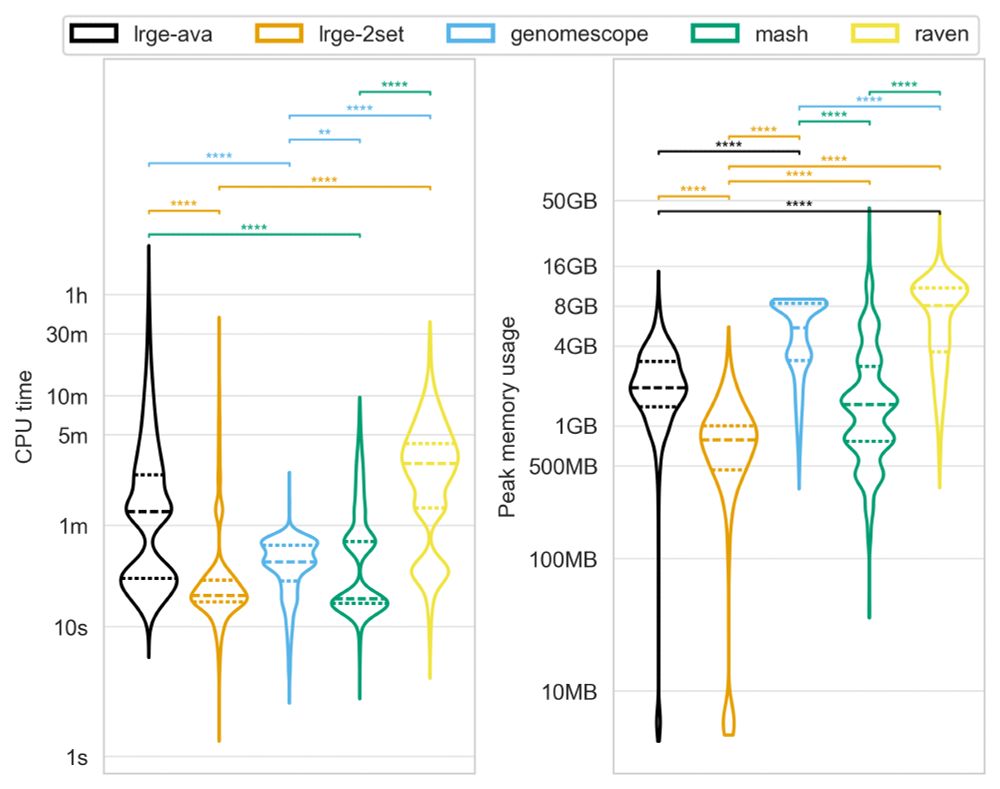
However, the computational resource usage (runtime/memory) of LRGE was MUCH better than assembling

November 7, 2025 at 3:19 AM
However, the computational resource usage (runtime/memory) of LRGE was MUCH better than assembling
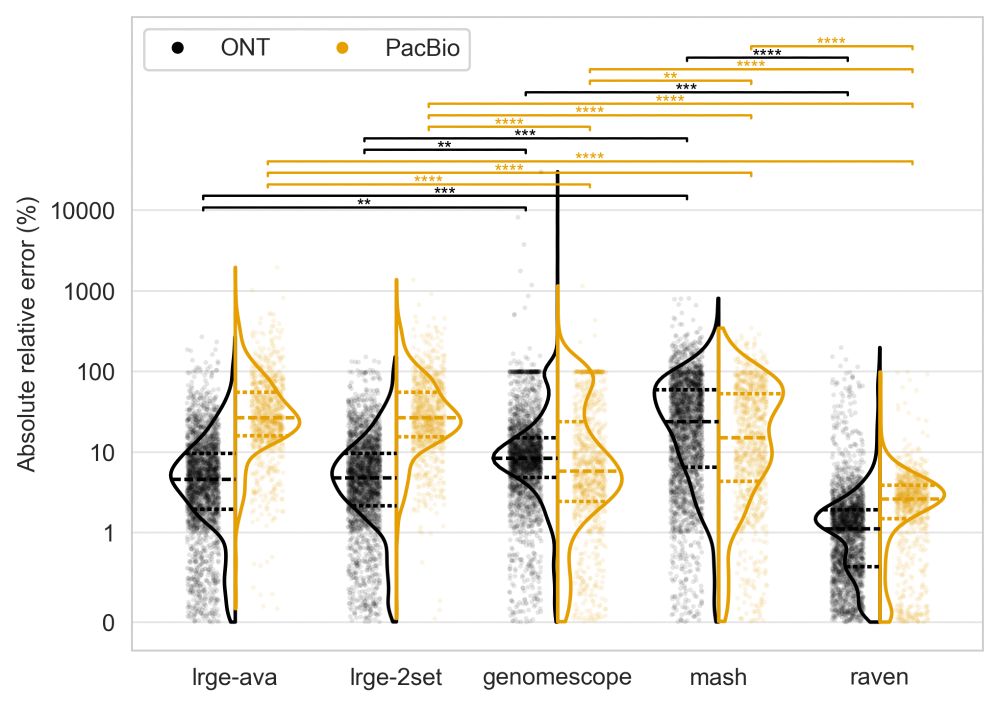
We benchmarked >3,000 bacterial genomes and found that LRGE (our method) achieves significantly better accuracy than k-mer-based methods like Mash and GenomeScope and performs on par with full genome assembly (Raven)

November 7, 2025 at 3:19 AM
We benchmarked >3,000 bacterial genomes and found that LRGE (our method) achieves significantly better accuracy than k-mer-based methods like Mash and GenomeScope and performs on par with full genome assembly (Raven)
The DOI URL doesn't seem to be working for the preprint currently. You can find it here: www.biorxiv.org/content/10.1...
www.biorxiv.org
December 3, 2024 at 4:02 AM
The DOI URL doesn't seem to be working for the preprint currently. You can find it here: www.biorxiv.org/content/10.1...
8/ Try it out!
LRGE is open-source and ready to integrate into your workflows as a Rust library or CLI application. Whether you’re on a high-performance cluster or a basic laptop, LRGE delivers fast and reliable genome size estimates. Get it here: github.com/mbhall88/lrge
LRGE is open-source and ready to integrate into your workflows as a Rust library or CLI application. Whether you’re on a high-performance cluster or a basic laptop, LRGE delivers fast and reliable genome size estimates. Get it here: github.com/mbhall88/lrge
GitHub - mbhall88/lrge: Genome size estimation from long read overlaps
Genome size estimation from long read overlaps. Contribute to mbhall88/lrge development by creating an account on GitHub.
github.com
December 3, 2024 at 1:38 AM
8/ Try it out!
LRGE is open-source and ready to integrate into your workflows as a Rust library or CLI application. Whether you’re on a high-performance cluster or a basic laptop, LRGE delivers fast and reliable genome size estimates. Get it here: github.com/mbhall88/lrge
LRGE is open-source and ready to integrate into your workflows as a Rust library or CLI application. Whether you’re on a high-performance cluster or a basic laptop, LRGE delivers fast and reliable genome size estimates. Get it here: github.com/mbhall88/lrge
7/ We validated LRGE on 3370 long read bacterial datasets which have associated high-quality RefSeq assemblies 🦠. We also confirmed it generalises to eukaryote organisms 🪰🌱🍞
December 3, 2024 at 1:38 AM
7/ We validated LRGE on 3370 long read bacterial datasets which have associated high-quality RefSeq assemblies 🦠. We also confirmed it generalises to eukaryote organisms 🪰🌱🍞
6/ And it’s efficient! ⚡
LRGE uses significantly less CPU and memory than traditional approaches, making it ideal for both high-performance clusters and resource-limited environments.
LRGE uses significantly less CPU and memory than traditional approaches, making it ideal for both high-performance clusters and resource-limited environments.
December 3, 2024 at 1:38 AM
6/ And it’s efficient! ⚡
LRGE uses significantly less CPU and memory than traditional approaches, making it ideal for both high-performance clusters and resource-limited environments.
LRGE uses significantly less CPU and memory than traditional approaches, making it ideal for both high-performance clusters and resource-limited environments.
5/ LRGE vs. the competition 🔥
LRGE delivers estimates as reliable as assembly-based methods and better than k-mer-based approaches.
Relative error (y-axis) measures the proportional difference between the estimated and true genome size.
LRGE delivers estimates as reliable as assembly-based methods and better than k-mer-based approaches.
Relative error (y-axis) measures the proportional difference between the estimated and true genome size.
December 3, 2024 at 1:38 AM
5/ LRGE vs. the competition 🔥
LRGE delivers estimates as reliable as assembly-based methods and better than k-mer-based approaches.
Relative error (y-axis) measures the proportional difference between the estimated and true genome size.
LRGE delivers estimates as reliable as assembly-based methods and better than k-mer-based approaches.
Relative error (y-axis) measures the proportional difference between the estimated and true genome size.
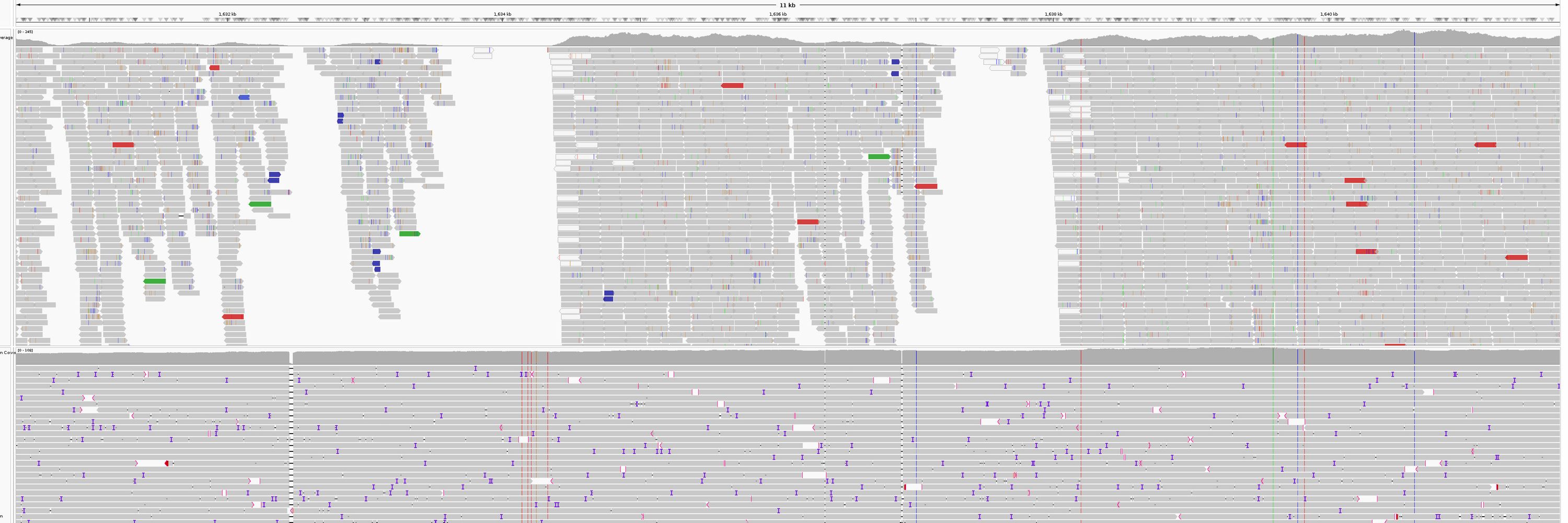
4/ LRGE also provides a confidence interval for the estimated genome size, offering users an expected range of variation.
December 3, 2024 at 1:38 AM
4/ LRGE also provides a confidence interval for the estimated genome size, offering users an expected range of variation.