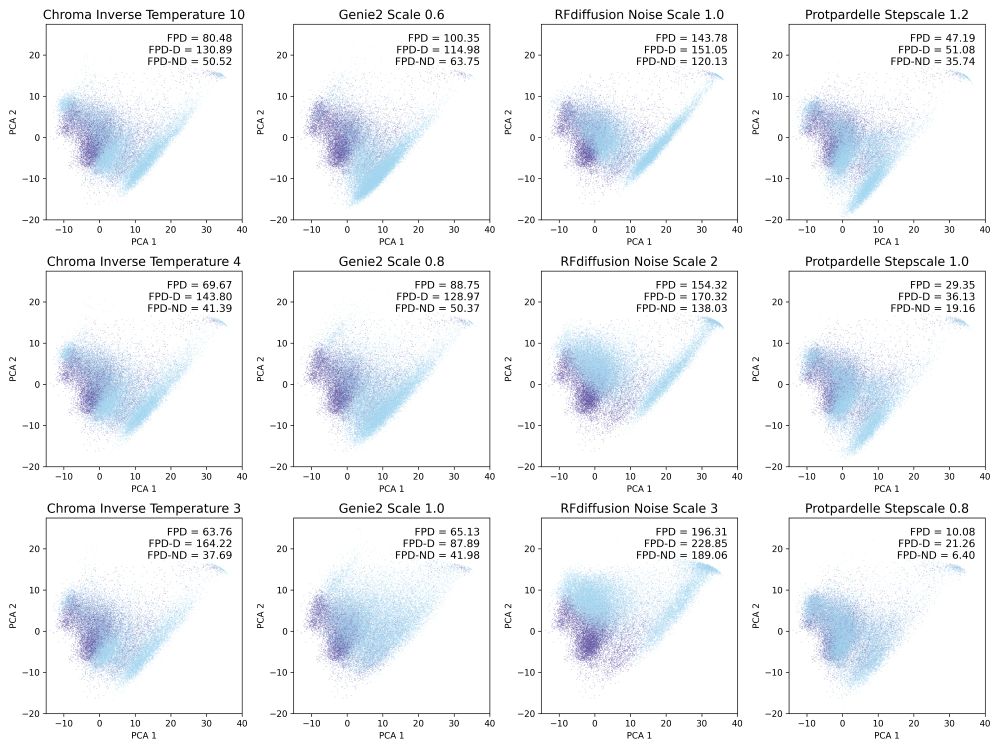

Possu Huang Lab
@possuhuanglab.bsky.social
470 followers
39 following
31 posts
Our lab uses experimental and computational methods to design de novo proteins | @Stanford
Posts
Media
Videos
Starter Packs
Pinned
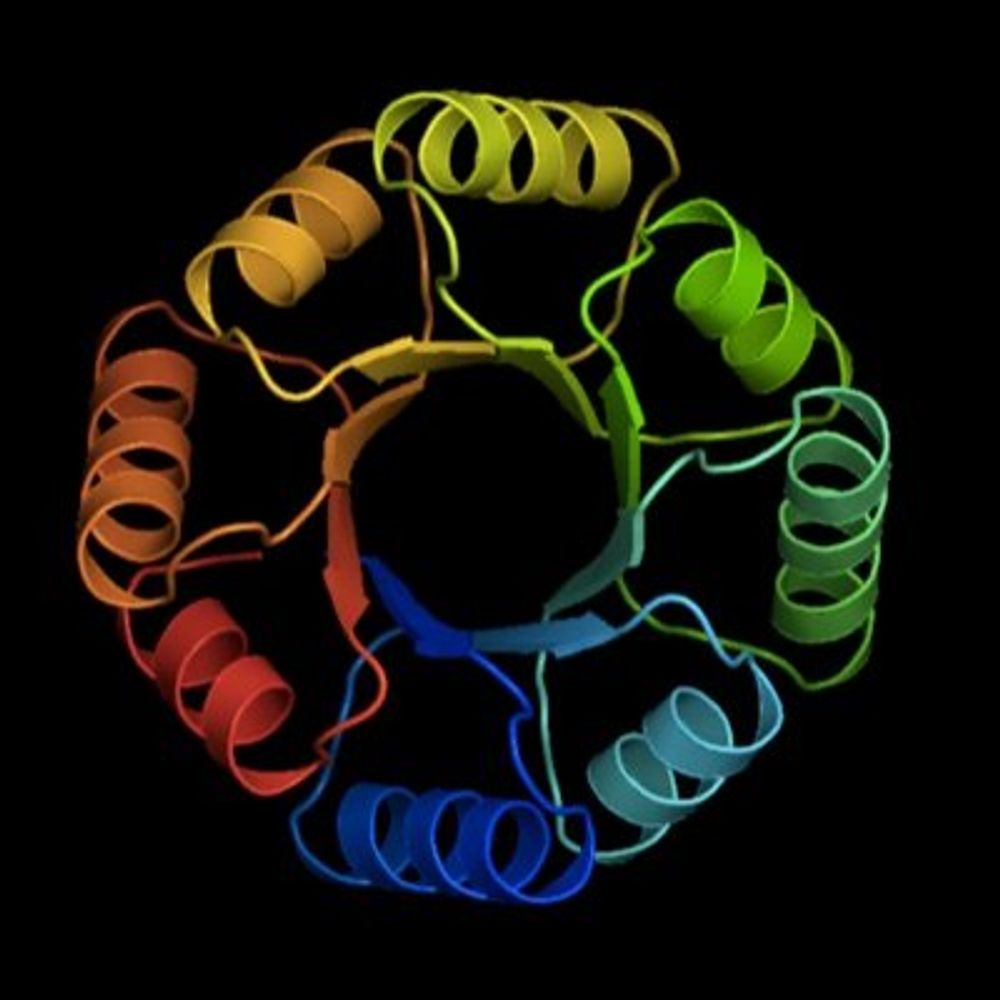
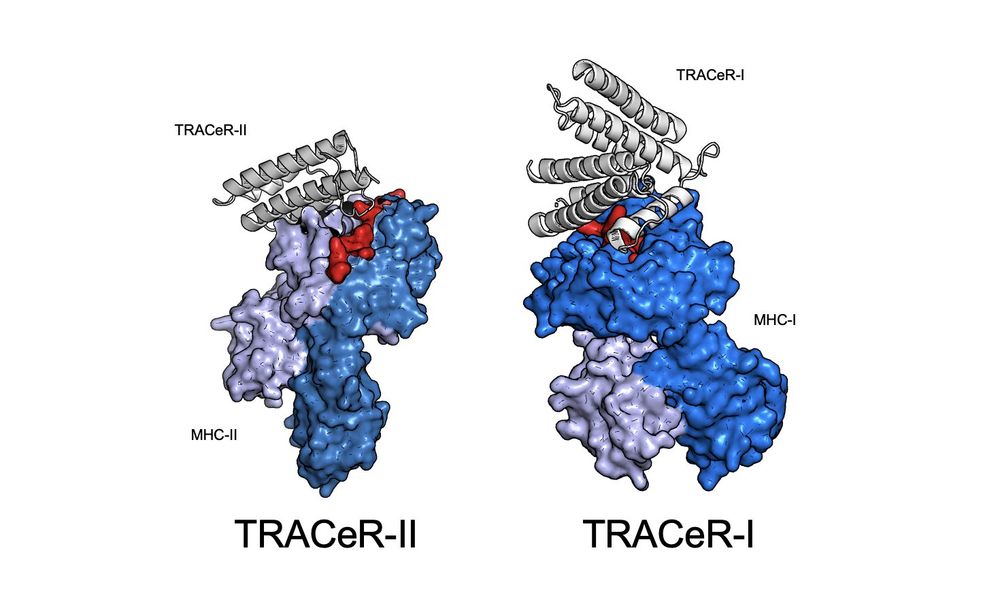









Reposted by Possu Huang Lab


Reposted by Possu Huang Lab